|
|
|
Medical Pharmacology Chapter 5: Autonomic Pharmacology: Adrenergic Drugs
|
|
|
|
|
|
|
(5) G protein and Second Messenger Systems:
Second messenger systems involve changes in concentration of molecules that themselves regulate downstream intracellular molecular events in response to initial receptor activation.
One of the primary second messenger systems involves changes in cAMP (cyclic AMP) concentration. Other second messenger systems include calcium ion and phosphoinositides.
Focusing on G protein systems: activation of receptors promotes a transition of a G protein from a relatively inactive state to an active state.
The activated form of G protein then changes the activity of another molecule, such as an enzyme or an ion channel state.
In the case of the enzyme, catalytic activity could be increased, for example. In the case of the ion channel, channel conductance (a measure of the ease by which the ion moves through the channel) is changed.
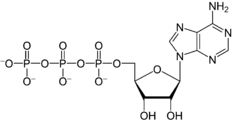
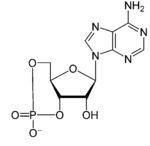
Considering cAMP, one enzyme that regulates cAMP levels is adenylyl cyclase, the activity of which is regulated by interaction with a G protein system. cAMP is formed by conversion of ATP to cAMP through a reaction catalyzed by adenylyl cyclase.
ATP → cAMP
adenylyl cyclase catalyzed reaction
Adenosine triphosphate (ATP)
cyclic AMP (cAMP)
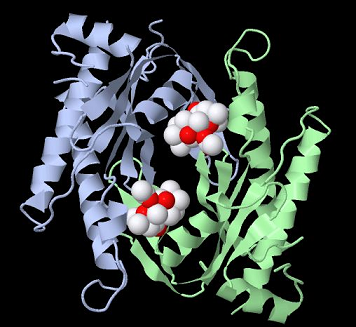
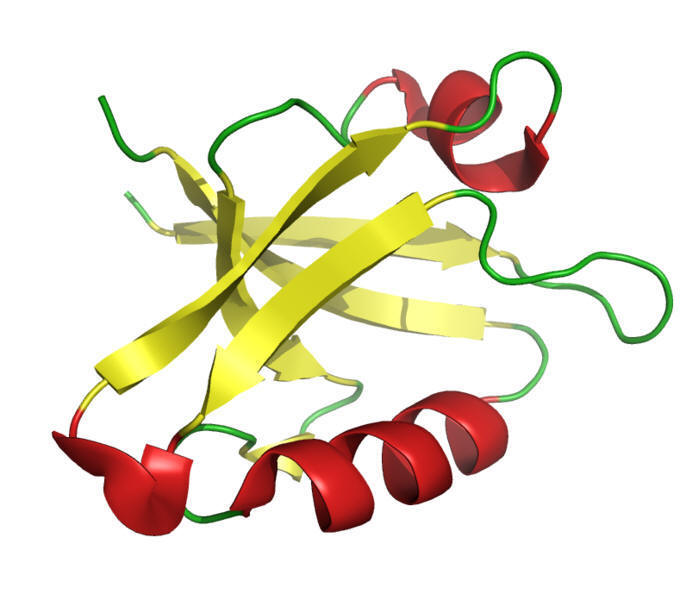
Type II adenylyl cyclase with two forskolin molecules bound31
Approximately 10 forms or isozymes of adenylyl cyclase have been identified in mammals.31
These isoforms have been designated adenylyl cyclase type I-X.
These forms exhibit some general characteristics.
For example, they exhibit two trans-membrane regions (M1,M2) consisting of six membrane-spanning helices; moreover, there are two cytoplasmic regions (C1,C2) which support not only catalysis but also regulation by G proteins and forskolin.
The above example is type II adenylyl cyclase.
Two associated C2 domains are bound together creating a homodimer required for ATP to cAMP and PPi catalysis.31,32,33
Binding of an adrenergic agonist to the adrenergic receptor results in a change in receptor form (conformational structural change) and promotes receptor-association with the membrane associated G protein complex and the subsequent reduction in affinity for GDP (guanosine diphosphate) by its G protein binding site.27
The net effect is a dissociation of GDP from G protein thus increasing the likelihood of GTP binding.
The GTP-bound G protein state is the active form.
Furthermore, also associated with GTP binding is a dissociation of one of the G protein subunits, the αs subunit from the βγ subunits.
This disassociation because of a reduction in affinity of GTP-αs for the βγ subunits.
Accordingly, hydrolysis of GTP back to GDP results in a tendency for the original G protein αsβγ configuration to be reestablished.
Depending on the system, the βγ subunit complex can also affect enzyme activity.
More generally the αs subunit can affect many systems, although we have focused on the adrenergic receptor.
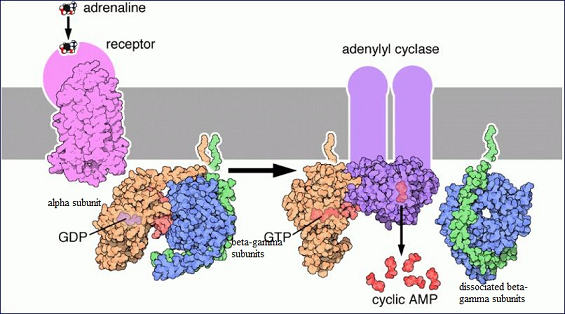
These steps are illustrated in the figure below:
Epinephrine-Receptor Association with Subsequent Activation of Adenylyl Cylcase via a G-Protein System32
In the illustration above, the adrenergic receptor is drawn in pink.
The G protein is composed of the αsβγ subunits (orange, blue, and green) and shows GDP association.
1. Following receptor activation by epinephrine (adrenaline), GDP dissociates and is replaced by GTP which results in activation of the GTP-αs -adenylyl cyclase complex which exhibits increased adenylyl cyclase activity.
2. This increase in enzyme activity causes an increase in intracellular cAMP concentration.
3. Also note that the βγ subunit has dissociated from αs.
4. The αsβγ structure is reestablished following GTP hydrolysis.
G Protein Receptor: Animation32a
α- and β-adrenergic receptors are coupled to "cytoplasmic scaffolding proteins" which themselves are associated with further downstream signaling systems.
One example: the β1-receptors interact with PZD proteins which are cytoskeletal elements.33 PZD is an acronym for first letters of three proteins:
(1) Post-synaptic density protein (PSD95),
(2) Zonula occludens-1 protein (zo-1). [the first 3 proteins found to exhibit a common "PZD domain."] and
(3) Drosophila disc large tumor suppressor (DIGA).
The PZD domain which is found in many signaling proteins is characterized by an 80-90 amino acid sequence which binds with a short region of the C-terminus of other proteins.
PDZ proteins exhibit a number of regulatory functions including coupling to adapter proteins, for example Grb2 or guanine-nucleotide exchange factors which regulate certain G proteins.9
|
|
Some G protein-coupled receptors can form functional dimers. β1 receptors can associate to form homodimers or β1 receptors can interact with β2 receptors to form heterodimers.
Furthermore, β1 receptors can also associate with α2 adrenergic receptors as well as δ-opiate receptors.9
These varied heterodimer associations may be a mechanism for mutual receptor regulation between receptor classes.
The major subclasses of α-receptors are α1- and α2 adrenergic receptors.
Activation of α1-receptors initiates a multi-steps signaling pathway beginning with G protein coupling.
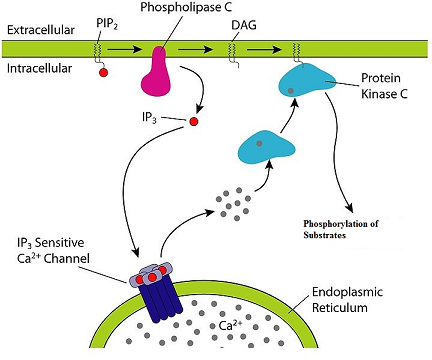
The interaction occurs principally with the G protein designated Gq which activates generation of IP3(inositol 1, 4-5-trisphosphate a.k.a triphosphoinositol).
Formation of IP3 is favored following Gq-activation of phospholipase C which hydrolyzes polyphosphoinositides.
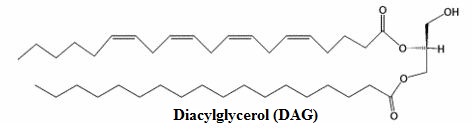
Along with inositol 1, 4-5-triphosphate, diacylglycerol (DAG) is also formed.
|
|
|
|
|
Increases in IP3 concentration promotes release of Ca2+ from intracellular sites thus increasing cytoplasmic free Ca2+.
Activation of this receptor system may also increase membrane conductance to Ca2+, causing an influx of extracellular Ca2+.
DAG increases the catalytic activity of protein kinase C (recall that kinases catalyze phosphorylation reactions, i.e. adding phosphoryl group).
Pathways that were historically identified with peptide growth factor receptor-activation of tyrosine kinases are also activated by α1-receptor stimulation; furthermore, α1-receptor activation also increases catalytic activity of both mitogen-activated kinases, a.k.a. MAP kinases and polyphosphoinositol-3-kinase (PI-3-kinase).
These latter two pathways may be associated with α1-adrenergic receptor regulation of cell proliferation.
|
|
Effects of α-receptor stimulation is dependent on the particular physiological system. 9
One example involves regulation of vascular smooth muscle tone.9
In that case, α1-receptor activation
(1) Increases Ca2+ intracellularly,
(2) Activates calmodulin thus promoting myosin light chain phosphorylation and
(3) Enhancing actin-myosin interactions. These effects promote smooth muscle contraction.
The receptor class is found also in intestinal smooth muscle, heart, liver, and in genitourinary tract smooth muscle.9
Receptors located in the genitourinary tract smooth muscle are targeted by α1-receptor antagonists (blockers) in the management of benign prostatic hypertrophy (BPH) which tends to reduce urinary flow.
These antagonists which work by competitive inhibition promote smooth muscle relaxation which is a reduction of normal sympathetic tone.
The development and application of these drugs have transitioned clinical management of BPH from surgery to pharmacological treatment. (In addition to the α1-adrenergic receptor blockers, 5α-reductase inhibitors are also used.)
Alfuzosin, doxazosin, tamsulosin, and terazosin are examples of selective α1-receptor antagonists used for BPH lower urinary tract symptoms (LUTS).
|
|
|
|
|
|
|
|
|
|
|
|
These agents work because α1-receptors are prevalent in prostate and bladder neck smooth muscle.37,38
There are at least 3 α1-adrenergic receptor subtypes: α1A, α1B, and α1D with the α1A subclass involved with smooth muscle tone in the prostate and bladder neck whereas α1B receptors regulate vascular smooth muscle contractility. α1D receptors may be associated with bladder muscle contraction and sacral spinal cord innervation.
Blockade of α1-adrenergic receptors in the prostate thus relaxes prostatic smooth muscle which improves urinary flow and other LUTS thus improving BPH-related bladder outlet obstruction.37,39
α2-adrenergic receptors when stimulated work through Gi which is an inhibitory G protein.9
Activation of Gi may have several signaling consequences including:
(1) adenylyl cyclase enzyme inhibition (inhibiting this enzyme decreases cAMP synthesis;
The βγ subunit complex may also contribute to adenylyl cyclase inhibition.)
(2) cell membrane hyperpolarization (more negative membrane potential, i.e. membrane potential moves farther away from threshold potential) by mechanism involving activation of the "inward rectifier" K+ ion channel and
(3) a reduction in Ca2+ channel conductance.
These effects reduce neurotransmitter release from the neuron so affected.
Recall that neurotransmitter release is a Ca2+-dependent process and typically requires membrane depolarization; therefore, reduced Ca2+ inward current and movement of the membrane resting potential away from threshold (more negative) are two events which reduce the likelihood of neurotransmitter released.
α2-receptors are localized both on the presynaptic side and the postsynaptic side.9
Activation of presynaptic α2-receptors reduces synaptic transmission; this effect is a type of feedback inhibition since neurotransmitters released by the presynaptic endings would not only activate postsynaptic receptors but by activating presynaptic receptors would inhibit additional transmitter release.9
Insulin release is inhibited by α2-adrenergic receptor activation on pancreatic beta cells.
Platelet aggregation is also suppressed by platelet α2-adrenergic receptor stimulation.
α2-receptor agonists acting in the central nervous system (CNS) decrease sympathetic outflow to peripheral sites, thus reducing norepinephrine-mediated increases in vascular smooth muscle tone.9
This effect is the basis for antihypertensive actions of α2-agonists.
Specific drugs used clinically in this way will be discussed later.
|
|
|
|
|
|
|
|
|
This Web-based pharmacology and disease-based integrated teaching site is based on reference materials, that are believed reliable and consistent with standards accepted at the time of development. Possibility of human error and on-going research and development in medical sciences do not allow assurance that the information contained herein is in every respect accurate or complete. Users should confirm the information contained herein with other sources. This site should only be considered as a teaching aid for undergraduate and graduate biomedical education and is intended only as a teaching site. Information contained here should not be used for patient management and should not be used as a substitute for consultation with practicing medical professionals. Users of this website should check the product information sheet included in the package of any drug they plan to administer to be certain that the information contained in this site is accurate and that changes have not been made in the recommended dose or in the contraindications for administration. Advertisements that appear on this site are not reviewed for content accuracy and it is the responsibility of users of this website to make individual assessments concerning this information. Medical or other information thus obtained should not be used as a substitute for consultation with practicing medical or scientific or other professionals. |