-
Occurs within large aqueous
components (e.g.,interstitial space, cytosol)
-
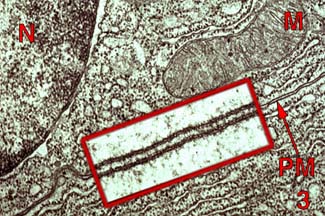
Occurs across epithelial membrane
tight junctions
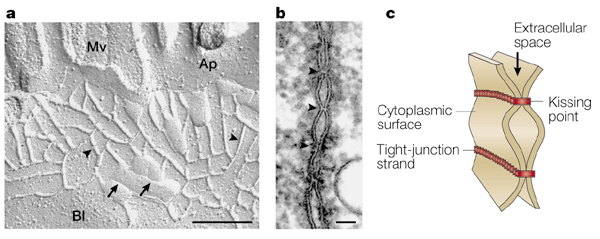
"Structure of tight junctions. a | Freeze-fracture replica electron microscopic image of intestinal epithelial cells. Tight junctions appear as a set of
continuous, anastomosing intramembranous particle strands or fibrils (arrowheads) on the P face with complementary vacant grooves on the E face
(arrows). (Mv, microvilli; Ap, apical membrane; Bl, basolateral membrane.) Scale bar, 200 nm. b | Ultrathin sectional view of tight junctions. At kissing points
of tight junctions (arrowheads), the intercellular space is obliterated. Scale bar, 50 nm. c | Schematic of
three-dimensional structure of tight junctions. Each tight-junction strand within a plasma membrane associates laterally with another tight-junction strand in the apposed membrane of an adjacent cell to form
a paired tight-junction strand, obliterating the intercellular space (kissing point)."--from
Nature Reviews Molecular Cell Biology 2; 285-293 (2001) MULTIFUNCTIONAL STRANDS IN TIGHT JUNCTIONS
Fick's
Law
|
Definition:
Fick's Law describes passive movement molecules
down its concentration gradient.
|
|
Flux (molecules
per unit time) = (C1 - C2) · (Area ·Permeability coefficient) /
Thickness
|
-
C1
is the higher concentration and C2
is the lower concentration
-
Area = area across which diffusion
occurs
-
Permeability coefficient: drug
mobility in the diffusion path
-
Thickness: length of the diffusion
path
Katzung, B. G. Basic Principles-Introduction ,
in Basic and Clinical Pharmacology, (Katzung, B. G., ed)
Appleton-Lange, 1998, p 5. |
|