|
|
|
|
|
|
|
Medical Pharmacology Chapter 16: Pharmacology of Antipsychotics Drugs
|
|
|
|
|
|
|
|
The discovery of chlorpromazine in the early 1950s represents one of the most consequential events in the history of psychiatry.
Prior to this era, patients with schizophrenia and other psychotic disorders were largely managed with sedation, physical restraints, or invasive procedures such as insulin coma therapy and frontal lobotomy.
![]() The introduction of the
first antipsychotic drug fundamentally altered that landscape.
The introduction of the
first antipsychotic drug fundamentally altered that landscape.
In 1952, Jean Delay and Pierre Deniker, a psychiatrist and neurologist working at the Hôpital Sainte-Anne in Paris, systematically evaluated the antihistamine promethazine derivative RPh-4560 (later named chlorpromazine) in psychotic patients.
Their seminal report, presented at the Congrès des Médecins Aliénistes et Neurologistes de France in 1952,1 described dramatic reductions in agitation and psychotic symptoms without rendering patients unconscious, a property they distinguished as a novel "neuroleptic" effect, from the Greek meaning "to clasp the neuron."
Haloperidol, synthesized by Paul Janssen in 1958 and introduced clinically in the early 1960s, offered greater potency and became the prototypic high-potency FGA.
Together, chlorpromazine and haloperidol defined the clinical and pharmacological profile of the firstgeneration (or "typical" or "conventional") antipsychotics, a class that would dominate schizophrenia pharmacotherapy for the next three decades.
The mechanistic basis of FGA efficacy was elucidated in the 1960s and 1970s through a series of landmark neurochemical studies.
![]() In 1963, Carlsson and Lindqvist demonstrated that administration of
chlorpromazine and haloperidol to mice increased the turnover of dopamine
metabolites (3-methoxytyramine and normetanephrine), suggesting that these
drugs were blocking postsynaptic dopamine receptors and triggering a
compensatory increase in presynaptic dopamine synthesis.2
In 1963, Carlsson and Lindqvist demonstrated that administration of
chlorpromazine and haloperidol to mice increased the turnover of dopamine
metabolites (3-methoxytyramine and normetanephrine), suggesting that these
drugs were blocking postsynaptic dopamine receptors and triggering a
compensatory increase in presynaptic dopamine synthesis.2
This observation laid the groundwork for the dopamine hypothesis of antipsychotic action.
In 1976, two landmark papers confirmed the central role of D2 dopamine receptors.
Creese, Burt, and Snyder demonstrated that the clinical potency of antipsychotic drugs correlated highly with their affinity for dopamine receptors as measured by radioligand binding assays.3
In the same year, Seeman and colleagues independently confirmed this finding using [3H]-haloperidol binding, establishing that neuroleptic dosing is directly proportional to D2 receptor affinity.4
![]() These
findings constituted the evidentiary foundation of the dopamine hypothesis:
that the antipsychotic efficacy of FGAs derives primarily from blockade of
D2 dopamine receptors, particularly in the mesolimbic pathway.
These
findings constituted the evidentiary foundation of the dopamine hypothesis:
that the antipsychotic efficacy of FGAs derives primarily from blockade of
D2 dopamine receptors, particularly in the mesolimbic pathway.
The brain contains four primary dopamine pathways, each mediating distinct physiological functions.
FGA blockade of D2 receptors in each of these pathways accounts for both the therapeutic effects and the adverse effect profile of this drug class:
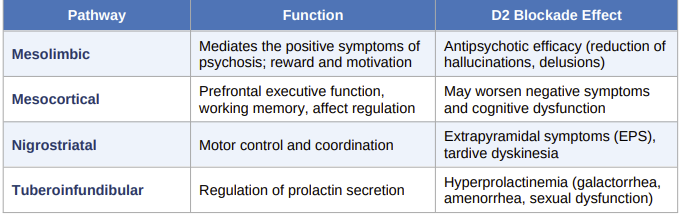 |
In addition to D2 antagonism, FGAs bind with varying affinity to histamine H1, muscarinic M1, and α-1 adrenergic receptors.
The clinical consequences of this off-target binding are significant and differ substantially between high- and low-potency agents:
Histamine H1 blockade
Sedation and weight gain.
More pronounced with low-potency agents such as chlorpromazine and thioridazine.
Muscarinic M1 blockade
Anticholinergic effects including dry mouth, urinary retention, constipation, blurred vision, and cognitive impairment. Most prominent with low-potency FGAs.
α-1 adrenergic blockade
Orthostatic hypotension, dizziness, and reflex tachycardia.
Also more common with low-potency agents.
Classification of First-Generation Antipsychotics
FGAs are classified by potency, defined by the milligram dose required to achieve an effect equivalent to 100 mg of chlorpromazine (the chlorpromazine equivalent, or CPZ-eq).
Potency correlates inversely with likelihood of sedation and anticholinergic effects, but directly with risk of extrapyramidal side effects.
|
|
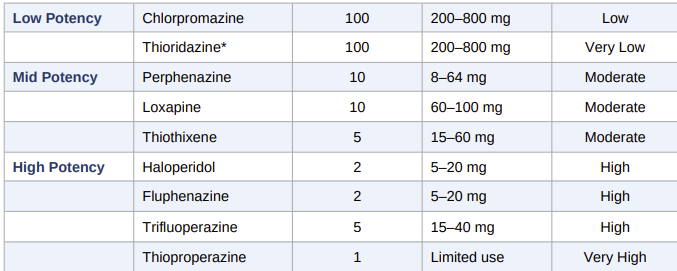 |
![]() A
clinically useful principle: high-potency FGAs trade sedation and
anticholinergic burden for a higher EPS risk, while low-potency agents carry
the opposite profile.
A
clinically useful principle: high-potency FGAs trade sedation and
anticholinergic burden for a higher EPS risk, while low-potency agents carry
the opposite profile.
Mid-potency agents such as perphenazine offer an intermediate balance and were specifically studied in the landmark CATIE trial as the FGA comparator.7
|
|
|
|
|
|
|
|
DISCLAIMER
|