|
|
|
|
|
|
|
Chapter 3: General Principles: Pharmacodynamics
|
|
|
|
|
|
|
![]() Pharmacodynamic
Variability in Special Populations
Pharmacodynamic
Variability in Special Populations
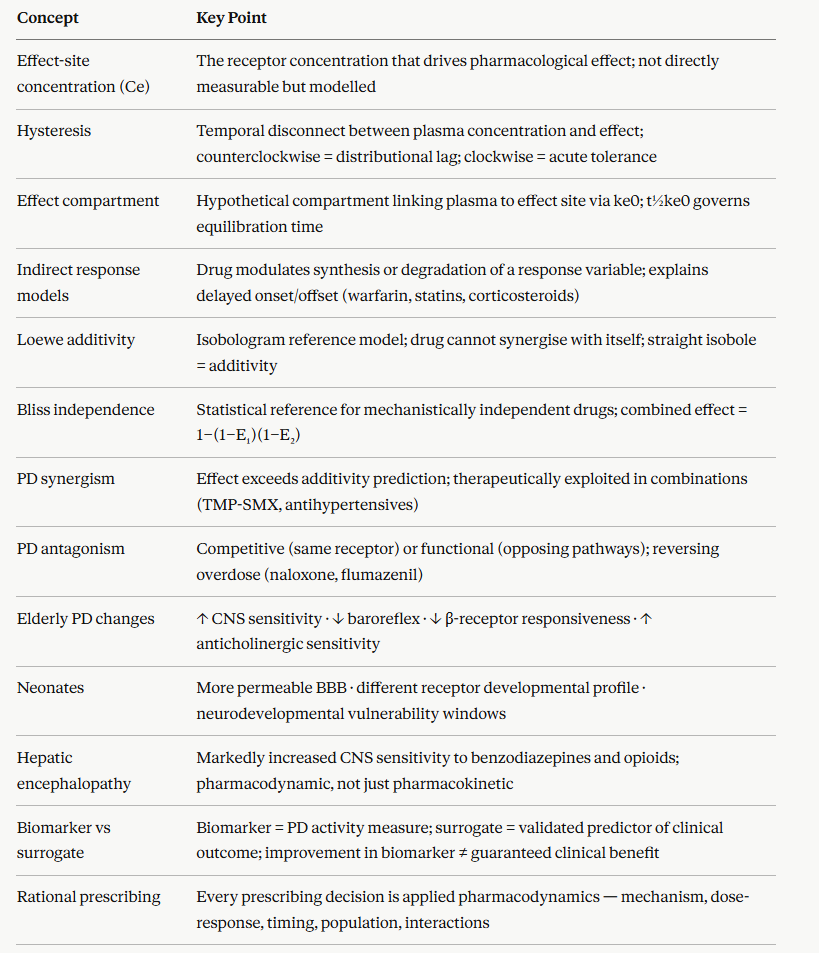
The concentration-effect relationship is not fixed as it varies systematically and predictably across identifiable patient subgroups.
Understanding these pharmacodynamic differences in special populations is essential for safe prescribing.1,2
Ageing produces pharmacodynamic changes that are independent of, and additive to, the well-recognized pharmacokinetic changes such as reduced renal clearance, reduced hepatic CYP activity, of old age:
CNS sensitivity to all sedating drugs (benzodiazepines, opioids, antihistamines, antipsychotics) is increased; the EC50 for sedation, cognitive impairment, and falls is lower in elderly patients at any given plasma concentration
Cardiovascular homeostatic reflexes are blunted as baroreflex sensitivity declines with age, which makes elderly patients disproportionately susceptible to orthostatic hypotension from vasodilator, antihypertensive, and antipsychotic medications.
β-Adrenoceptor responsiveness decreases with age affecting both chronotropic and inotropic responses to β-agonists are reduced.
This reduction appears partly due to receptor downregulation and altered G protein coupling efficiency
Sensitivity to anticholinergic effects such as confusion, urinary retention, constipation is increased as a result of reduced cognitive reserve and reduced cholinergic neurotransmission associative with aged brain.1,2
Neonates and pediatric patients
Pharmacodynamic development is as important as pharmacokinetic maturation in neonatal and pediatric pharmacology.
The neonatal blood-brain barrier is more permeable than in adults; CNS drug penetration (and CNS toxicity) is greater for many drugs at equivalent plasma concentrations.
Opioid receptor populations and G protein coupling efficiencies differ in neonates, producing both altered analgesic responses and altered side effect profiles.
Neurodevelopmental windows of vulnerability exist during which certain drug exposures e.g anesthetic agents and benzodiazepines, may produce lasting effects on brain development.
Pregnancy alters pharmacodynamics through several mechanisms, with direct clinical implications.
Progesterone reduces lower oesophageal sphincter tone, increasing aspiration risk with sedating drugs.
Uterine sensitivity to oxytocin and its antagonists changes progressively through gestation, requiring dose adjustment for tocolytic agents.
Seizure threshold changes in pregnancy affect antiepileptic drug pharmacodynamics.
Hepatic and renal failure alter pharmacodynamics beyond their pharmacokinetic consequences.
In hepatic encephalopathy, brain sensitivity to benzodiazepines and opioids is markedly increased with patients exhibit exaggerated CNS depression at standard plasma concentrations.
This CNS response appears partly due to upregulation of GABA-A receptors and accumulation of endogenous benzodiazepine-like substances.
In chronic renal failure, uremic toxins alter drug-receptor interactions and ion channel function, contributing to altered CNS sensitivity independent of drug accumulation.
Heart failure alters tissue perfusion and receptor sensitivity, β-adrenoceptor downregulation in the failing heart reduces the inotropic response to catecholamines and β-agonists, explaining part of the clinical picture of end-stage cardiac failure1,2
Biomarkers and Surrogate Endpoints in Pharmacodynamic Assessment
A challenge in clinical pharmacodynamics is measuring the effect-site pharmacodynamic response in a patient.
For many drugs, the ultimate clinical endpoint such as prevention of myocardial infarction or prevention of cancer progression or survival, is too distant in time and too confounded by other variables to serve as practical pharmacodynamic information during dose optimization or drug development.
Biomarkers may be of help as they are measurable biological variables whose change reflects drug pharmacodynamic activity, even if they are not themselves the clinical endpoint.
Distinction between biomarker types matters clinically:4
A pharmacodynamic biomarker directly reflects receptor or pathway activity.
The biomarker changes immediately and proportionally with drug concentration-effect.
![]() INR in
warfarin therapy, troponin in response to myocardial injury, LDL-C
in response to statins, CD4 count in HIV therapy, HbA1c in
diabetes management, and QTc prolongation in drug cardiac safety
studies are all pharmacodynamic biomarkers.
INR in
warfarin therapy, troponin in response to myocardial injury, LDL-C
in response to statins, CD4 count in HIV therapy, HbA1c in
diabetes management, and QTc prolongation in drug cardiac safety
studies are all pharmacodynamic biomarkers.
They are useful for dose adjustment and evidence of drug activity.
A surrogate endpoint is a biomarker that is reasonably expected to predict clinical benefit or harm, validated against the hard clinical outcome.
LDL-C reduction is a validated surrogate for cardiovascular event prevention, supported by outcome trial data.
Tumor shrinkage is an accepted surrogate for survival in many cancer trials.
Surrogacy requires explicit validation.
A biomarker that improves under treatment does not automatically indicate that patients will do better.
There are examples of drugs that improved biomarkers while worsening outcomes.
For example, the antiarrhythmic agents encainide/flecainide in CAST, where suppression of ventricular ectopy as a biomarker led to worse mortality.4
Translating the PK-PD Framework to Rational Prescribing
The eight modules of this chapter converge on a single practical principle: rational prescribing is applied pharmacodynamics.
Every clinical prescribing decision can be understood as a decision about receptor pharmacology, and the PK-PD framework provides the language to make that reasoning explicit.
A prescriber who understands the material in this chapter can, for any drug:
Predict the time to peak effect from knowledge of the effect-site equilibration half-life
Anticipate the duration of effect even after plasma concentrations have fallen
Explain tolerance and dose escalation requirements from receptor regulation principles
Predict adverse effects from off-target receptor binding profiles
Adjust doses rationally in special populations based on known pharmacodynamic changes
Interpret therapeutic drug monitoring concentrations in the context of the full concentration-effect relationship
Combine drugs rationally, exploiting synergism or avoiding dangerous potentiation
Recognize the difference between pharmacokinetic and pharmacodynamic drug failure
|
|
|
|
|
|
|
|