|
|
|
|
|
|
|
Chapter 3: General Principles: Pharmacodynamics
|
|
|
|
|
|
|
Pharmacodynamics is the science of what drugs do to the body.
At its most fundamental level, that question reduces to a smaller one: how does a drug molecule interact with a specific molecular target in the body to produce a biological effect?
The answer, for the overwhelming majority of drugs, lies in the receptor concept based on the idea that drugs produce their effects by binding selectively and reversibly to specific protein molecules in cells, and that the magnitude of the effect is a predictable function of that binding interaction.
Receptor theory is not merely background knowledge for pharmacodynamics rather it is pharmacodynamics.
Every clinical phenomenon that will be explored in this module: why some drugs are more potent than others, why partial agonists have ceiling effects, why tolerance develops, why drug interactions occur at the receptor level follows logically and mathematically from the principles established in this module.
Mastering receptor theory first is therefore not optional preamble; it is the conceptual foundation on which everything else is built.1,2
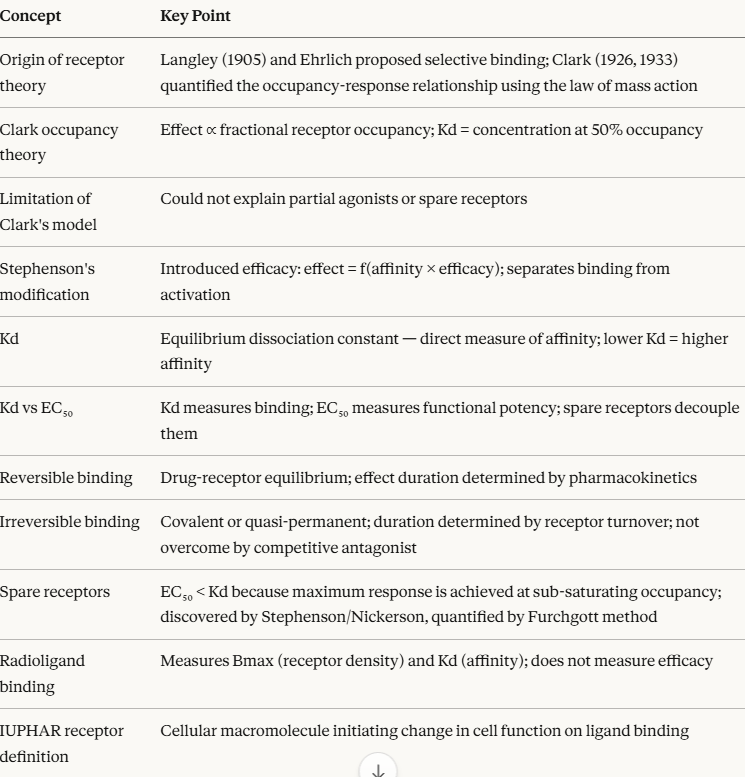
Receptor Concept: Historical Foundations
The receptor concept has a precise origin in intellectual history, and understanding that history illuminates why the concept is formulated the way it is.
Langley and Ehrlich (1900–1910) independently proposed that cells must possess specific chemical structures capable of selectively recognizing and binding to exogenous compounds.
John Newport Langley, working with nicotine and curare at the neuromuscular junction, proposed the existence of a "receptive substance" on the surface of muscle cells in 1905 lrepresenting a structure that could be activated by nicotine and blocked by curare.
Working in parallel, Paul Ehrlich proposed his side-chain theory of cell surface receptors, which provided the conceptual basis for his famous Zauberkugel (magic bullet) concept of selective pharmacological targeting.1
Alfred Joseph Clark (1885–1941) transformed these qualitative concepts into quantitative science. Working with acetylcholine on isolated muscle preparations, Clark was the first to demonstrate that the relationship between drug concentration and biological effect follows a mathematical function derived from the law of mass action.3
His pivotal 1926 paper established what we now call the receptor occupancy theory — the hypothesis that:
1. Drug molecules combine reversibly with specific receptors according to the law of mass action.
2. The biological effect is directly proportional to the fraction of receptors occupied by the drug.
3. The maximum effect is achieved when all receptors are occupied.
Clark's occupancy theory introduced the two most fundamental parameters in quantitative pharmacodynamics: affinity (how tightly a drug binds to its receptor) and the concept that drug effects can be quantitatively related to its concentration through a mathematically defined curve.3,4
The limitation of Clark's model became apparent when experimental data repeatedly violated its central assumption: that maximal receptor occupancy always produces maximal effect.
Two observations proved troubling:
1. Some drugs produced maximal responses while occupying only a fraction of the total receptor population (spare receptors).
2. Some compounds that clearly bound to receptors could produce only a submaximal response regardless of concentration (what we now call partial agonists).
Clark's model had no mechanism to explain either phenomenon.
Ariëns (1954) and Stephenson (1956) independently solved this problem by separating receptor occupancy from receptor activation.
Ariëns introduced the concept of intrinsic activity representing a proportionality constant between receptor occupancy and the biological response it produced.
Stephenson's more rigorous modification introduced the concept of efficacy which measures the capacity of a drug-receptor complex to produce a stimulus in the tissue, independent of the drug's affinity for the receptor.5
His approah established that a drug's action at a receptor is the product of two independent properties:
how well it binds (affinity) and
what it does once bound (efficacy).
An antagonist has affinity but zero efficacy.
A full agonist has both high affinity and high efficacy.
A partial agonist has affinity but submaximal efficacy.
This framework will be developed in full in a module to follow but we introduce it here, at the beginning.5,10
Mass Action and Equilibrium Binding
The quantitative foundation of receptor pharmacology is the law of mass action, which describes the reversible interaction between a drug (ligand, L) and its receptor (R) to form a drug-receptor complex (LR): L ↔ LR.
At equilibrium, the rates of complex formation and dissociation are equal. This equilibrium is characterized by the equilibrium dissociation constant (Kd), defined as the ratio of the dissociation rate constant to the association rate constant: Kd = k_off / k_on
This is a critical insight: the Kd is a direct measure of affinity.
A drug with a Kd of 1 nM binds with far greater affinity than one with a Kd of 1 µM.
The drug with a Kd of 1 nM it occupies 50% of receptors at a concentration 1,000 times lower than the drug with the Kd of 1µM.
From the law of mass action, the fractional receptor occupancy (the proportion of receptors occupied by drug at any given concentration) is described by:
Occupancy = [L] / ([L] + Kd)
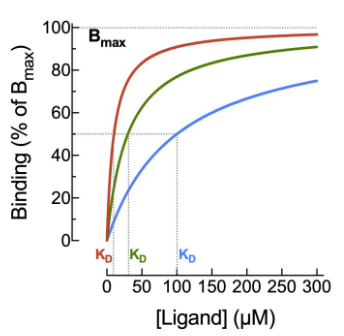
This equation, mathematically equivalent to the Hill-Langmuir equation and Clark's occupancy equation, produces a hyperbolic curve when plotted on a linear concentration scale,
Attribution:
Holt A 2023 Creative Commons.
https://pressbooks.openeducationalberta.ca/abcofpkpd/chapter/hyperbola/
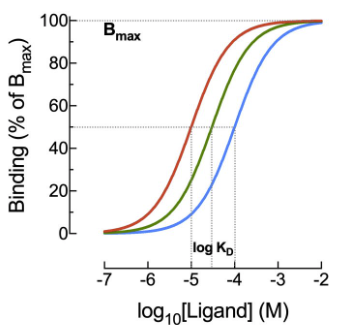
and the familiar sigmoid curve when plotted on a log concentration scale.
Attribution:
Holt A 2023 Creative Commons.
https://pressbooks.openeducationalberta.ca/abcofpkpd/chapter/hyperbola/
Several features of this equation deserve explicit attention:
When [L] = Kd: occupancy = 50% — this is the definition of Kd
When [L] ≈ Kd: occupancy ≈ [L]/Kd, occupancy is approximately proportional to concentration (linear region)
When [L] ﹥ Kd: occupancy approaches 100%, the receptor population is saturated
The sigmoid log-concentration-occupancy curve is one of the most important graphs in all of pharmacology, and it will recur throughout this chapter in many forms.
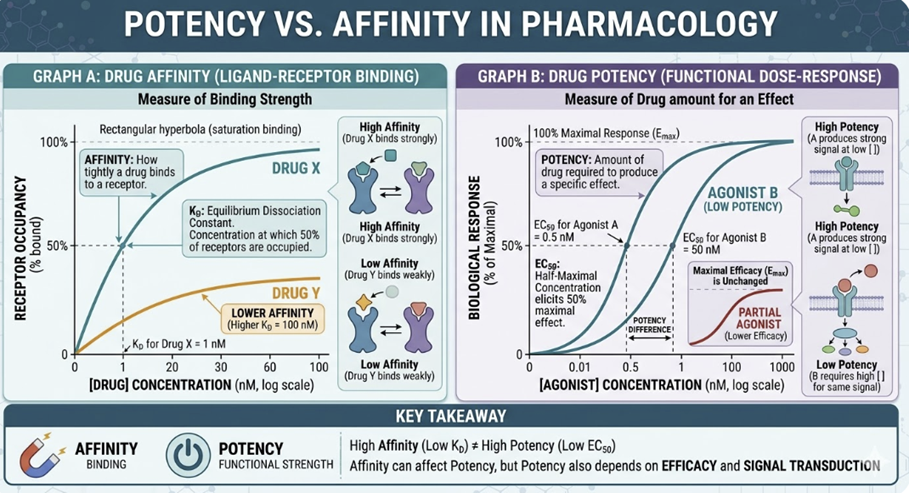
Affinity ,a binding property, quantified by Kd, is sometimes confused with potency, a functional property, quantified by EC50 which represents the drug concentration producing 50% of maximum effect.
These terms are related but distinct.
For a pure agonist with no spare receptors, Kd ≈ EC50
However, spare receptors fundamentally decouple affinity from potency.
Therefore, a drug can have a lower potency than its affinity would predict if few spare receptors are present, or a much higher potency if large receptor reserve exists.
The distinction between Kd and EC50 is one of the most clinically important concepts in receptor theory.7,8
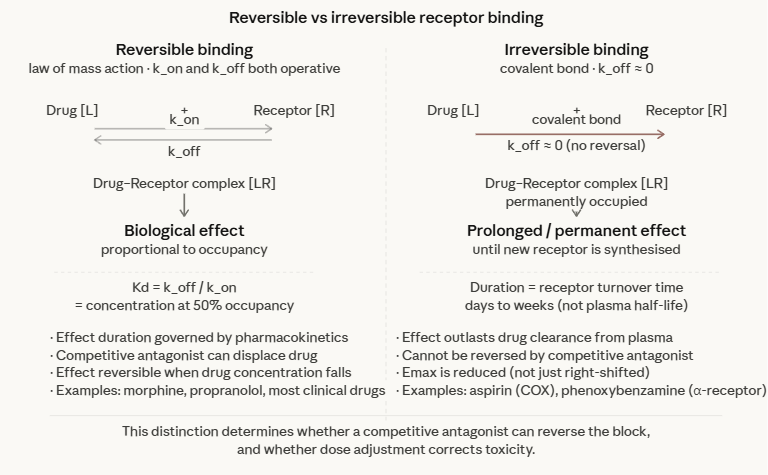
Reversible vs. Irreversible Binding
Reversible Binding
The law of mass action as formulated above describes reversible binding as the condition in which the drug-receptor interaction is in continuous dynamic equilibrium, with the drug associating and dissociating continually, as affected by drug concentration at the receptor sites.
The duration of drug effect is directly related to the time the drug maintains therapeutic concentrations at the receptor.
The effect can be terminated by removing the drug or by competitive displacement with an antagonist.
The relationship between drug concentration and effect is predictable and reversible.
Irreversible binding
Irreversible binding occurs when a drug forms a covalent bond with its receptor, or when the dissociation rate constant (koff) is so low that the drug-receptor complex is effectively permanent on the timescale of clinical relevance. (The off rate (koff)) describes how slow or fast the drug dissociates from the receptor).
In the "irreversible" case, the off rate is very, very low.
The clinical consequences are fundamentally different:
The duration of effect is determined not by pharmacokinetics but by the time required to synthesize new receptor protein — typically days to weeks
The effect cannot be overcome by reducing plasma drug concentration
The effect cannot be reversed by a competitive antagonist
Aspirin irreversibly acetylates the active site of cyclooxygenase (COX-1 and COX-2), which is why its antiplatelet effect persists for the full lifespan of the platelet (7–10 days) long after the drug has been eliminated.
Phenoxybenzamine, an irreversible α-adrenergic antagonist, is used in pheochromocytoma precisely because its prolonged receptor blockade protects against catecholamine surges.
Organophosphate nerve agents and pesticides irreversibly inhibit acetylcholinesterase by forming a stable phosphoryl-enzyme complex, and produce effects that usually persist until new enzyme is synthesized.1,2
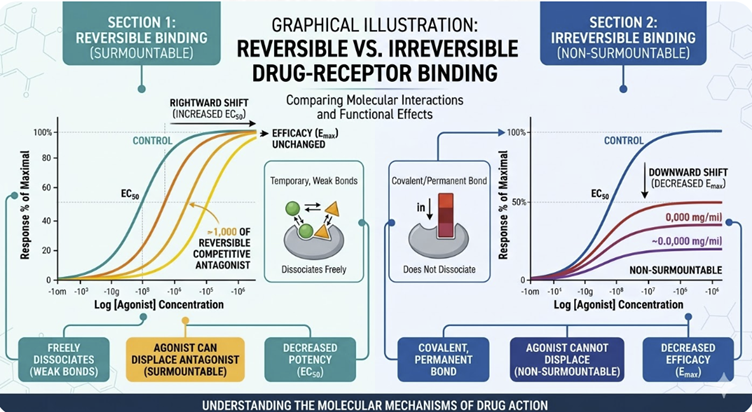
The distinction between reversible and irreversible binding also matters for the interpretation of dose-response curves.
An irreversible antagonist reduces the maximum achievable response (Emax) even at high agonist concentrations and no amount of additional agonist can overcome the block.
A reversible competitive antagonist, by contrast, shifts the dose-response curve to the right without reducing Emax, because sufficient agonist can always compete off the antagonist. This distinction will be examined in detail in a later section.
Spare Receptors and Receptor Reserve
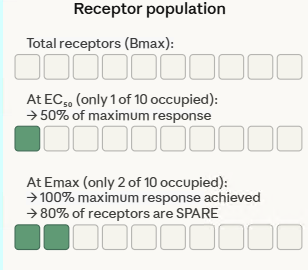
One of the most counter-intuitive discoveries in receptor pharmacology was that many tissues contain far more receptors than are needed to produce a maximum biological response.
This concept, spare receptors or receptor reserve, was first identified for histamine receptors in guinea pig ileum, where Stephenson and Nickerson demonstrated that occupancy of only 1–2% of available receptors was sufficient to elicit a maximal contractile response.5,6
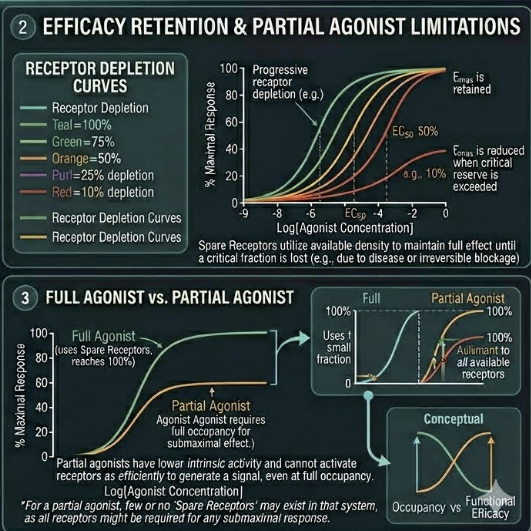
Furchgott provided the experimental methodology to quantify receptor reserve rigorously, using irreversible receptor antagonists to progressively inactivate portions of the receptor population.⁶ The key observations were:
(1) Irreversible inactivation of a substantial fraction of receptors reduced agonist potency (shifted the EC50 to higher concentrations) but did not reduce Emax thus the maximum response was preserved
(2) Only after inactivating a sufficiently large fraction of receptors did Emax begin to fall, revealing the point at which the receptor reserve had been exhausted
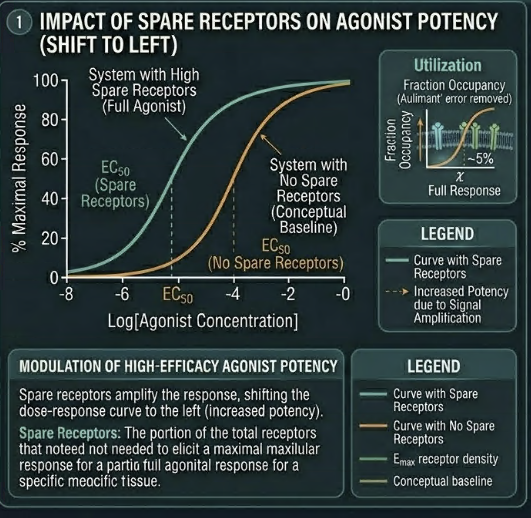
First, spare receptors amplify the apparent potency of high-efficacy agonists.
Because only a small fraction of receptors need be occupied to produce a maximum response, the concentration of agonist required to produce EC₅₀ (half-maximum effect) is much lower than the Kd (the concentration required to occupy 50% of receptors).
This is why the EC50 of a high-efficacy agonist can be far lower than its Kd , the pharmacokinetic measurement of plasma concentration significantly overestimates the concentration needed at the receptor for full effect.
Second, spare receptors explain why drugs can maintain therapeutic efficacy even when receptor numbers are reduced by disease, desensitization, or pharmacological blockade, up to the point at which receptor reserve is exhausted. This has direct implications for tolerance to be discussed later.
Third, the existence of spare receptors explains why partial agonists cannot achieve maximum system response even at saturating concentrations.
A partial agonist, by definition, generates less stimulus per receptor occupied than a full agonist.
When spare receptor reserve is large, a partial agonist may still achieve near-maximal response at low concentrations.
But as receptor number falls (through desensitization or disease), the partial agonist's intrinsic limitation becomes clinically manifest as a reduced ceiling effect.7.9
Measuring Receptor Binding: Radioligand Binding Assays
The receptor concepts described above were initially developed through functional pharmacology, observing biological responses to drugs in isolated tissues.
The direct measurement of drug-receptor binding became possible with the development of radioligand binding assays, which remain the primary experimental method for characterizing receptor-drug interactions in vitro.
In a radioligand binding assay, a radiolabelled drug (typically ³H- or ¹²⁵I-labelled) with known high affinity for the receptor of interest is incubated with a receptor-containing preparation (membrane fragments, intact cells, or purified protein).
After allowing equilibration, bound and free radioligand are separated (typically by filtration or centrifugation), and the radioactivity in the bound fraction is measured.
Saturation binding experiments use increasing concentrations of radioligand to characterize two key parameters:
Bmax represents the total receptor density (maximum binding capacity), expressed as fmol/mg protein or number of binding sites per cell.
Kd is the the equilibrium dissociation constant, determined as the radioligand concentration at which 50% of receptors are occupied.
Competition binding experiments use a fixed concentration of radioligand and increasing concentrations of an unlabelled competitor to determine the competitor's affinity for the receptor.
The concentration of competitor that displaces 50% of the radioligand defines the Ki (inhibition constant), which can be converted to Kd using the Cheng-Prusoff equation.8
Radioligand binding data has several important limitations that students must understand. Binding assays measure affinity that is, how tightly a drug interacts with a receptor but provide no direct information about efficacy.
Agonists, antagonists, and inverse agonists with similar affinities may be indistinguishable in a binding assay, because all three occupy the receptor.
Only functional assays measuring biological responses can distinguish them.
In living cells, the receptor may exist in multiple conformational states, may be coupled to G proteins or other signalling molecules, and may be subject to allosteric modulation.
These factors may change the apparent affinity measured functionally.
Working Definition of a Receptor
The IUPHAR Committee on Receptor Nomenclature provides the authoritative working definition:
"A receptor is a cellular macromolecule, or an assembly of macromolecules, that is concerned directly and specifically in chemical signaling between and within cells. Combination of a hormone, neurotransmitter, drug, or intracellular messenger with its receptor(s) initiates a change in cell function."8
Several aspects of this definition deserve emphasis.
First, a receptor is defined functionally by its role in chemical signaling not purely structurally.
Simple binding sites on proteins that do not initiate a change in cell function are not classified as receptors in the pharmacological sense.
Second, the definition encompasses intracellular receptors (nuclear hormone receptors) as well as cell surface receptors.
Third, the definition is explicitly pluralistic given that many receptors function as assemblies of multiple subunits (e.g., GABA-A receptors, nicotinic acetylcholine receptors), and receptor pharmacology must account for the properties of the assembled complex, not just individual subunits.
In clinical pharmacology, the receptor concept provides the framework for understanding not just how a drug works, but:
why it works
why it produces its specific combination of therapeutic effects and adverse effects
why it requires a specific dose, and
why its effects change under specific clinical conditions.
Every prescribing decision is, at its most fundamental level, a decision about receptor pharmacology.
Summary
|
|
|
|
|
|
|
|