|
|
|
|
|
|
|
Chapter 3: General Principles: Pharmacodynamics
|
|
|
|
|
|
|
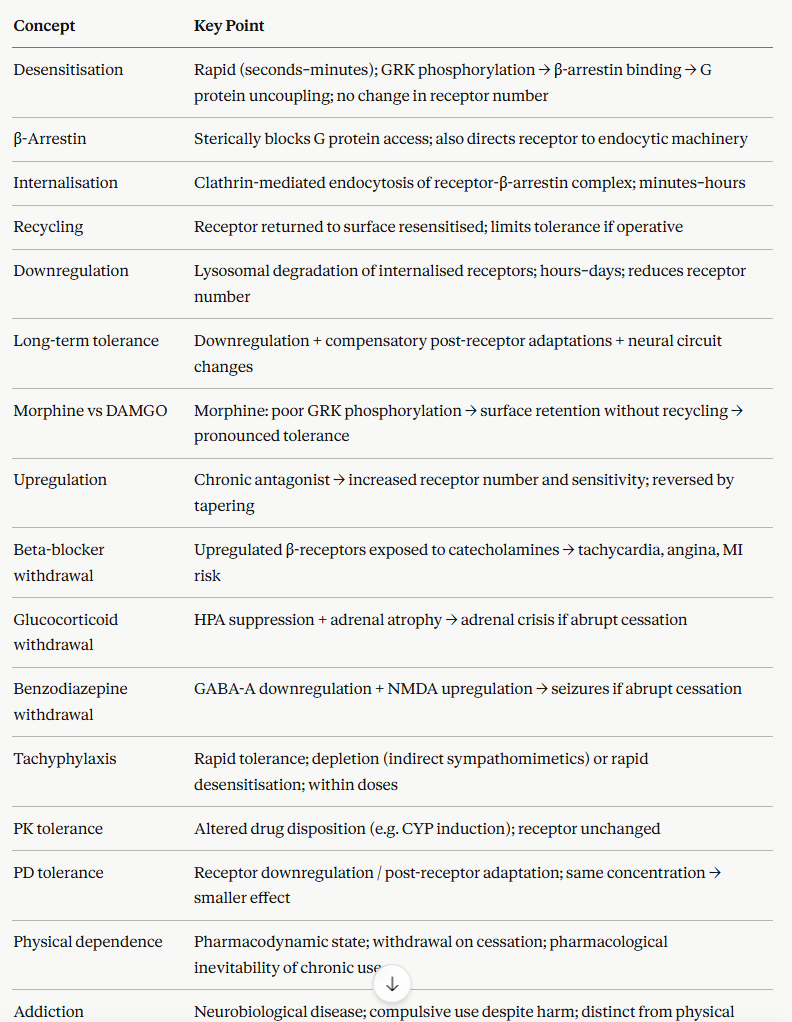
Pharmacodynamic vs Pharmacokinetic Tolerance: A Critical Distinction
Tolerance, the requirement for a higher dose to achieve the same effect, can arise through two entirely different mechanisms, and distinguishing them is clinically important because they require different management strategies.
Pharmacokinetic tolerance results from altered drug disposition over time, such that the same dose produces a lower plasma concentration.
The most common mechanism is CYP enzyme induction in which chronic exposure to the drug or a co-administered inducer up-regulates hepatic cytochrome P450 (CYP enzymes), accelerating the drug metabolism.
Carbamazepine, rifampicin, and phenobarbital are all potent CYP inducers that produce significant pharmacokinetic auto-induction and their own plasma concentrations fall with continued use at a fixed dose.
Under these circumstance dose must be increased to maintain therapeutic levels.
The tolerance is at the pharmacokinetic level; receptor sensitivity is unchanged.
![]() Pharmacodynamic
tolerance results from receptor-level changes.
Pharmacodynamic
tolerance results from receptor-level changes.
These changes include desensitization, down-regulation, or compensatory post-receptor adaptations such that the same concentration of drug at the receptor produces a smaller effect than initially.
Drug pharmacokinetics may be entirely unchanged as tolerance is due to changes at the receptor end of the concentration-effect relationship.
Opioid analgesic tolerance is predominantly pharmacodynamic.
Plasma morphine concentrations rise appropriately with dose escalation, but receptor down-regulation and compensatory cellular adaptations mean that higher concentrations are required to achieve the same analgesic effect.1,2,5,6
![]() Combined
tolerance is common.
Combined
tolerance is common.
For example in chronic alcohol use, ethanol induces hepatic CYP2E1, representing pharmacokinetic tolerance with faster ethanol metabolism, while simultaneously producing GABA-A receptor down-regulation and NMDA receptor up-regulation representing pharmacodynamic tolerance manifest as reduced CNS ethanol sensitivity.
Both mechanisms must be recognized when managing alcohol use disorder.
Physical Dependence vs Addiction
Physical dependence and addiction are frequently conflated in clinical and public discourse, but they are pharmacologically distinct phenomena with different clinical implications.
Physical dependence is a pharmacodynamic state.
Physical dependence is defined by the presence of a withdrawal syndrome on drug discontinuation or dose reduction.
Such dependences stems from an automatic physiological consequence of prolonged receptor regulation (upregulation or compensatory adaptation) and occurs:
when certain drugs are administered chronically and at sufficient dose independent of existing addiction or psychological disorder.
Patients who take therapeutic opioids for chronic pain, long-term benzodiazepines for anxiety, chronic corticosteroids for inflammatory disease, or antidepressants for depression are likely to develop some degree of physical dependence.
Management consists of gradual tapering.1,2
Addiction (substance use disorder) is a neurobiological disease characterised by compulsive drug seeking and use despite harmful consequences.
Addiction is driven by dysregulation of dopaminergic reward circuitry and executive function.
![]() Addiction
involves craving, loss of control, and continued use
despite harm and these features that are not predicted
by or required for physical dependence.
Addiction
involves craving, loss of control, and continued use
despite harm and these features that are not predicted
by or required for physical dependence.
Physical dependence does not cause addiction, and addiction can occur without significant physical dependence (as with cocaine or some stimulants).
Failure to distinguish these concepts has direct clinical harms: under-treatment of pain in physically dependent patients due to misidentification of dependence as addiction, and under-recognition of addiction in patients who have no obvious withdrawal syndrome.1,2
|