Medical Pharmacology Chapter 2 General Principles: Pharmacogenomics
|
|
|
|
|
|
|
|
-
-
Audio Overview: Pharmacogenomics (Extended)
Audio Overview: Pharmacogenomics (Brief)
-
-
Introduction: From Population Pharmacology to Individualized Prescribing
-
Classical pharmacology describes drug effects in populations resulting in average responses at average doses in average patients.
-
But no patient is average.
-
Substantial inter-individual variability in drug response has always been observed clinically, and for most of pharmacology's history that variability was attributed to differences in age, organ function, body composition, and co-medications.
-
-
-
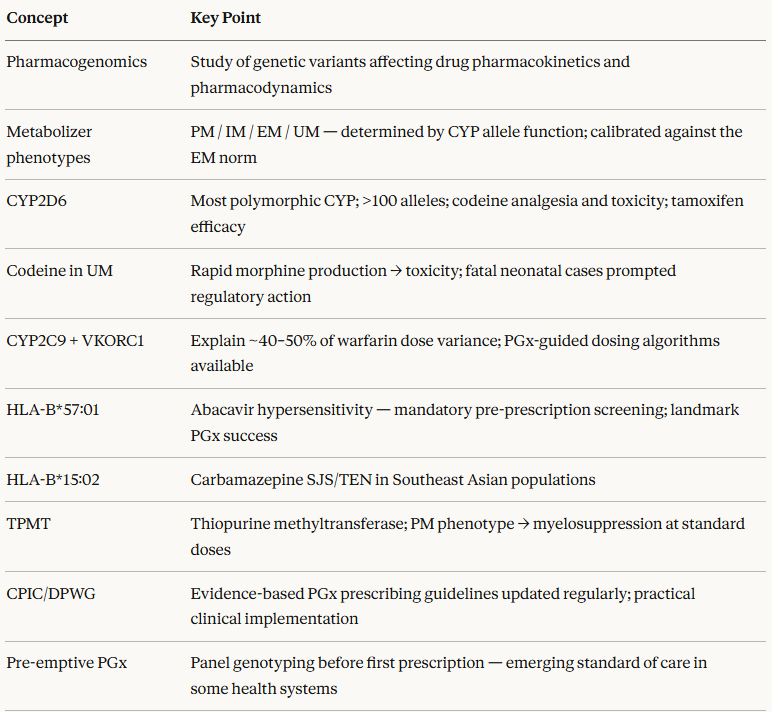
 Pharmacogenomics,
the study of how genetic variation influences drug response, provides
deeper insights into sources of this variability.
Pharmacogenomics,
the study of how genetic variation influences drug response, provides
deeper insights into sources of this variability.
-
Inherited differences in drug-metabolizing enzymes, drug transporters, and drug targets alter the pharmacokinetics and pharmacodynamics of drugs.
-
Some of these differences lead to an understanding of the clinical consequences in ways that may medical benefits to the patient.
-
-
The promise of pharmacogenomics is prescribing precision which entails, using personalized genetic information, selecting the right drug at the right dose for that patient.1,2
-
-
-
-
Genetic Polymorphisms: The Molecular Basis of Variable Drug Response
-
A genetic polymorphism is a variation in DNA sequence that occurs in more than 1% of the population. Polymorphisms relevant to pharmacogenomics occur in three categories of genes:3
-
Drug-metabolizing enzyme genes
-
These variants that alter CYP enzyme function, affecting the rate at which drugs are metabolized and therefore their plasma concentrations and duration of action.
-
-
-
These variants in genes encoding membrane transporters (P-glycoprotein, organic anion transporters, SLCO/OATP transporters) that alter drug absorption, distribution, and excretion.
-
-
-
These variants in receptor, ion channel, or enzyme genes that alter the pharmacodynamic response to a drug at a given concentration.
-
The clinical consequences of these polymorphisms are expressed as metabolizing phenotypes which is the functional classification of an individual's drug-metabolizing capacity based on their genotype.
-
-
-
-
-
-
The CYP enzyme system displays clinically important genetic polymorphisms, particularly in CYP2D6, CYP2C9, and CYP2C19.
-
These enzymes are responsible for the metabolism of a large fraction of commonly prescribed drugs.
-
Four metabolizer phenotypes are recognized:3,4
-
-
Carry two non-functional alleles.
-
Drug metabolism via the affected CYP isoform is absent or severely impaired.
-
Substrates accumulate to high concentrations which may result in toxicity at standard doses or manifest as an inability to convert prodrugs to active metabolites.
-
-
Intermediate Metabolizers (IM)
-
Carry one functional and one reduced-function allele.
-
Drug metabolism is reduced compared to normal exhibiting intermediate plasma concentrations and effects.
-
-
Extensive (Normal) Metabolizers (EM)
-
Carry two functional alleles.
-
Standard drug metabolism representing population "norm" against which standard drug doses are calibrated.
-
-
-
Carry multiple copies of a functional allele (gene duplication), typically for CYP2D6.
-
Drug metabolism is dramatically accelerated resulting plasma drug concentrations that are very low at standard doses.
-
These very low drug levels may result in inadequate therapeutic effect.
-
For prodrugs, conversion to active metabolites may be dangerously rapid.
-
-
-
-
Frequency
of these phenotypes varies significantly by ethnic
population, a consideration of practical importance
in diverse clinical populations.4
-
-
-
-
-
CYP2D6: The Most Clinically Important Pharmacogenomic Enzyme
-
CYP2D6 metabolizes approximately 20–25% of commonly used drugs and displays the most extensive pharmacogenomic variation of any CYP enzyme, with over 100 identified alleles, ranging from null (no function) to highly amplified (ultrarapid activity).
-
CYP2D6 is not inducible by drugs or dietary factors with its activity is determined almost entirely by genotype.5
-
Structure of CYP2D6 
-
Attribution
-
BorisTM, Public domain, via Wikimedia Commons
-
https://commons.wikimedia.org/wiki/File:CYP2D6_structure.png March 14, 2006
-
-
-
-
Codeine and the CYP2D6 PM/UM spectrum
-
Clinical consequences of CYP2D6 polymorphism are apparent when describing codeine metabolism.
-
Codeine is a prodrug requiring O-demethylation by CYP2D6 to morphine for analgesic activity.
-
-
In poor metabolizers: CYP2D6 activity is absent or negligible.
-
Codeine is not converted to morphine.
-
The patient receives no analgesia.
-
This
circumstance represents the pharmacogenomic basis for
inter-individual variability in analgesic efficacy
associated with codeine administration.
-
-
-
In ultrarapid metabolizers: CYP2D6 converts codeine to morphine far more rapidly and completely than anticipated.
-
At standard doses, plasma morphine concentrations reach levels that in a normal metabolizer would require much higher doses.
-
 Deaths have been reported in ultrarapid metabolizer neonates
breastfed by ultrarapid metabolizer mothers prescribed
codeine postpartum.
Deaths have been reported in ultrarapid metabolizer neonates
breastfed by ultrarapid metabolizer mothers prescribed
codeine postpartum.-
These cases prompted regulatory restrictions on codeine use in pediatrics and nursing mothers in multiple countries.5
-
-
-
-
-
-
Tamoxifen, the cornerstone of adjuvant treatment for estrogen receptor-positive breast cancer, is metabolized by CYP2D6 to endoxifen, its most pharmacologically active metabolite.
-
Endoxifen exhibits about 100-fold greater affinity for the estrogen receptor compared to tamoxifen.
-
In CYP2D6 poor metabolizers, endoxifen plasma
concentrations are dramatically reduced, correlating in
some studies with worse clinical outcomes.
-
This
pharmacogenomic interaction also means that CYP2D6
inhibitors co-prescribed with tamoxifen, such as
fluoxetine or paroxetine, both potent CYP2D6 inhibitors
often prescribed for hot flushes associated with
tamoxifen therapy, may pharmacologically convert an
extensive metabolizer into a phenotypic poor
metabolizer.4
-
-
-
-
-
-
CYP2C9 and Warfarin: The Benchmark Pharmacogenomic Interaction
-
The vitamin K antagonist warfarin has become one of the benchmark drugs for pharmacogenomics.
-
Two factors appear responsible for this designation:
-
(1) Its narrow therapeutic indes and
-
(2) Because two genes together explain a large fraction of the variance in stable therapeutic dose requirements across individuals.6
-
-
-
CYP2C9:
-
The principal enzyme responsible for metabolism of the more pharmacologically active S-enantiomer of warfarin.
-
The CYP2C92 and CYP2C93 alleles, which are among the most common pharmacogenomic variants in European populations, encode enzymes with substantially reduced activity.
-
Carriers require lower warfarin doses to avoid supratherapeutic anticoagulation and have a significantly elevated risk of serious bleeding complications, particularly during dose initiation.
-
-
-
VKORC1
-
The gene encoding Vitamin K Oxide Reductase Complex subunit 1 which is the pharmacodynamic target of warfarin.
-
VKORC1 polymorphisms alter the sensitivity of the target enzyme, with certain variants producing a VKORC1 enzyme that is more easily inhibited by warfarin (lower dose required) or less easily inhibited (higher dose required).
-
VKORC1 polymorphisms and CYP2C9 polymorphisms together explain approximately 40–50% of the variance in stable warfarin dose requirements, a level of pharmacogenomic predictability that has driven the development of dosing algorithms incorporating genotype alongside clinical variables (age, body surface area, co-medications).6
-
-
-
-
HLA Alleles and Immune-Mediated Adverse Reactions
-
Pharmacogenomics extends beyond drug metabolism to the pharmacodynamics of immune-mediated adverse reactions.
-
Certain severe Type B adverse drug reactions are strongly associated with specific HLA alleles, allowing prospective identification of at-risk patients.1,7
-
-
Abacavir hypersensitivity syndrome (HLA-B*57:01)
Abacavir, a nucleoside reverse transcriptase inhibitor used in HIV treatment, causes a potentially life-threatening hypersensitivity syndrome in approximately 5–8% of patients.
-
The reaction is almost exclusively confined to carriers of the HLA-B57:01 allele. Prospective HLA-B57:01 screening before abacavir initiation has essentially eliminated abacavir hypersensitivity syndrome in practice.
-
The FDA label requires HLA-B*57:01 testing before prescribing abacavir.
-
This represents one of the most successful translations of pharmacogenomic knowledge into clinical practice.7
-
-
-
-
Carbamazepine and Stevens-Johnson Syndrome (HLA-B15:02, HLA-A31:01)
-
Carbamazepine causes Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), life-threatening severe cutaneous adverse reactions, at greatly elevated frequency in carriers of HLA-B15:02 (predominantly Southeast Asian populations).
-
HLA-A31:01 is associated with carbamazepine hypersensitivity reactions across multiple ethnic groups.
-
Many regulatory authorities in affected populations now require HLA screening before carbamazepine initiation.
-
-
-
Allopurinol and SJS/TEN (HLA-B*58:01)
-
The gout medication allopurinol causes severe cutaneous reactions at elevated frequency in HLA-B58:01 carriers.
-
Mandatory pre-prescription screening has been implemented in several Asian countries with high HLA-B58:01 allele frequency.
-
-
-
-
-
Thiopurine methyltransferase (TPMT) is the enzyme responsible for metabolizing thiopurine drugs such as azathioprine, mercaptopurine, and thioguanine.
-
These drugs are used as immunosuppressants and in the treatment of childhood leukaemia.
-
TPMT genetic polymorphisms have direct, well-established clinical consequences.8
-
-
-
Approximately 1 in 300 individuals is a TPMT poor metabolizer (homozygous for non-functional alleles); approximately 10% of the population is heterozygous (intermediate metabolizer).
-
In poor metabolizers, the thiopurine drugs cannot be
adequately metabolized and accumulate as cytotoxic
thioguanine nucleotides in bone marrow, causing
life-threatening myelosuppression at standard doses.
-
-
Pre-treatment TPMT genotyping (or phenotypic enzyme activity testing) allows dose reduction in intermediate metabolizers and avoidance of thiopurine drugs in poor metabolisers in favour of alternative immunosuppressive strategies.
-
This approach is likely appropriate when prescribing azathioprine or mercaptopurine.9
-
-
-
-

|
|
|
|
|
|
References
|
|