Medical Pharmacology Chapter 1: General Principles: Overview and Introduction
|
|
|
|
|
|
|
Every time a drug is administered, the body immediately begins working on it.
Four sequential but overlapping processes, Absorption, Distribution, Metabolism, and Excretion (ADME), determine three things that matter most in the clinical setting:
How much drug actually reaches the site of action
How quickly it gets there
How long it stays at therapeutic concentrations
Understanding these processes conceptually is not optional background knowledge since it is the foundation for every dosing decision, every route-of-administration choice, and every anticipation of drug interactions.
A drug with perfect pharmacodynamic potency at its receptor is clinically useless if pharmacokinetics prevents it from reaching that receptor at an adequate concentration.1,2
The four ADME processes do not occur in strict sequence.
They overlap, interact, and begin simultaneously almost as soon as a drug enters the body.
However, we will consider them in order.
Absorption is the process by which a drug moves from its site of administration into the systemic circulation.
Absorption is the first and often most consequential pharmacokinetic step, because it determines whether any drug reaches the body at all.3
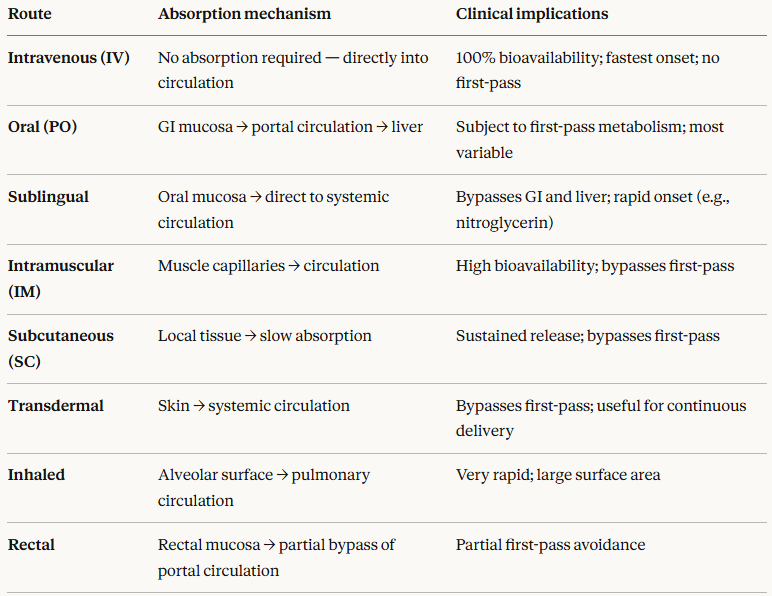
Route of administration is the single most important determinant of absorption. The major routes and their clinical implications are:
Bioavailability is the key concept arising from absorption.
Bioavailabiltiy is defined as the fraction of an administered dose that reaches the systemic circulation in an active (unchanged) form.
By definition, intravenous administration has 100% bioavailability.4
All other routes yield bioavailability less than 100%, due to incomplete absorption, chemical degradation in the GI tract, or first-pass metabolism.
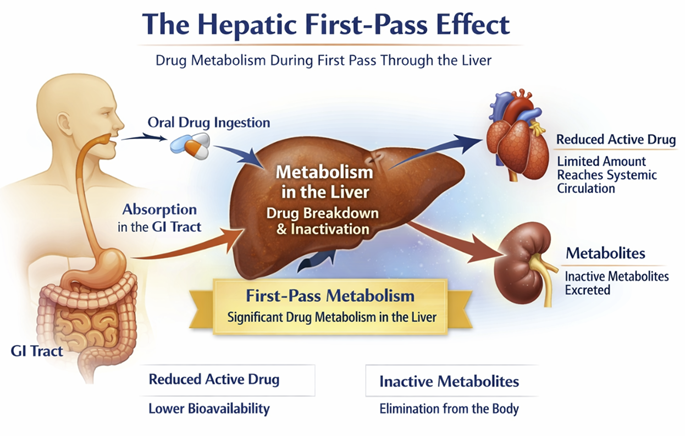
The First Pass Effect (Why Oral Dosing May Be Challenging)
When a drug is swallowed, it is absorbed through the gut wall and carried via the portal vein directly to the liver before reaching the systemic circulation.
The liver, the body's primary metabolic organ, may convert a substantial proportion of the drug into inactive metabolites before it ever has a chance to reach its target.
This hepatic first-pass effect (also called presystemic metabolism) can dramatically reduce the bioavailability of orally administered drugs.5
Classic examples of the first-pass effect
Nitroglycerin is almost completely destroyed by hepatic first-pass metabolism when swallowed; it must be given under the tongue (sublingually) or as a patch (transdermally) to be effective.
Morphine undergoes extensive first-pass metabolism, requiring that oral doses must be 2–3 times higher than intravenous doses to achieve equivalent effect.
Propranolol is well absorbed from the GI tract but loses approximately 75% of its active dose to first-pass hepatic metabolism.
When switching a patient from IV to oral administration of such a drug (or vice versa), the dose must be adjusted accordingly.
Failure to do so is a recognized source of medication errors.5
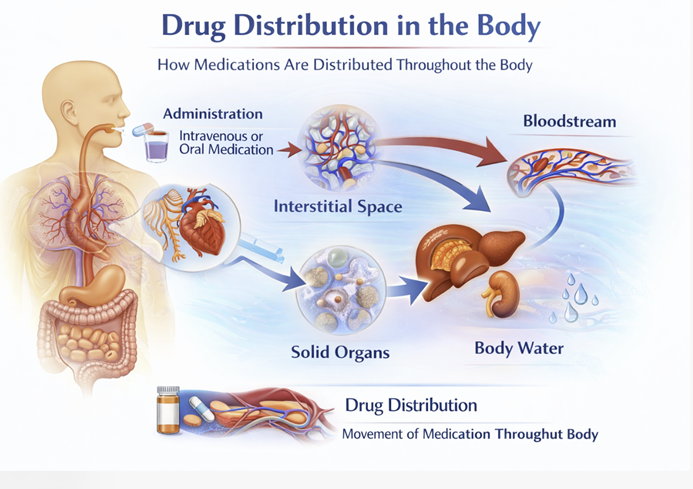
Once a drug enters systemic circulation, it distributes from the blood into tissues.
Distribution describes this transition from blood to tissue targets, but it is not uniform.
Different drugs partition into different tissues in dramatically different proportions, and this has major clinical consequences.6
Several factors govern how a drug distributes
Lipid solubility is perhaps the most important.
Lipophilic (fat-soluble) drugs readily cross cell membranes and accumulate in fat tissue and the brain.
Hydrophilic (water-soluble) drugs tend to remain in the blood and extracellular fluid and penetrate poorly into the CNS.
Plasma protein binding has a profound influence on distribution.
Drugs in the bloodstream exist in two forms: bound to plasma proteins (primarily albumin and α-1-acid glycoprotein) and unbound (free).
Only the free fraction can leave the circulation, cross cell membranes, and reach the site of action.
Only the free fraction is available for metabolism and excretion.7
This principle has immediate clinical relevance.
In patients with hypoalbuminemia (malnutrition, liver disease, nephrotic syndrome), the free fraction of highly protein-bound drugs is elevated, potentially causing toxicity at standard doses.
Phenytoin, warfarin, and many drugs that have narrow-therapeutic-indicies are particularly affected.
Special barriers restrict drug entry into certain compartments.
The most clinically important is the blood-brain barrier (BBB), formed by tight junctions between cerebral capillary endothelial cells.
Only lipophilic, un-ionized drugs of low molecular weight readily cross the BBB. This is why some antibiotics (e.g., many penicillins) achieve poor CNS penetration, while others are specifically designed to cross it.
The placenta is another critical barrier as most drugs do cross it to some degree, which is the basis for teratogenicity concerns in pregnancy.
The volume of distribution (Vd) is the pharmacokinetic parameter that captures where a drug goes in the body.
Conceptually, it represents the apparent volume of fluid that would be required to contain all the drug in the body at the same concentration measured in plasma.
A very large Vd (e.g., amiodarone: ~60 L/kg) means the drug distributes extensively into tissues and is largely absent from plasma.
A small Vd (e.g., warfarin: ~0.14 L/kg) means the drug stays mostly in the circulation, tightly bound to plasma proteins.
Detailed calculation of Vd and its clinical applications are in the Pharmacokinetics chapter.
Metabolism is the biochemical modification of a drug by the body.
The liver is the primary site of drug metabolism, though the gut wall, lungs, kidneys, and plasma also contribute.
The overall goal of metabolism is to convert lipophilic compounds into more water-soluble forms that can be excreted.8
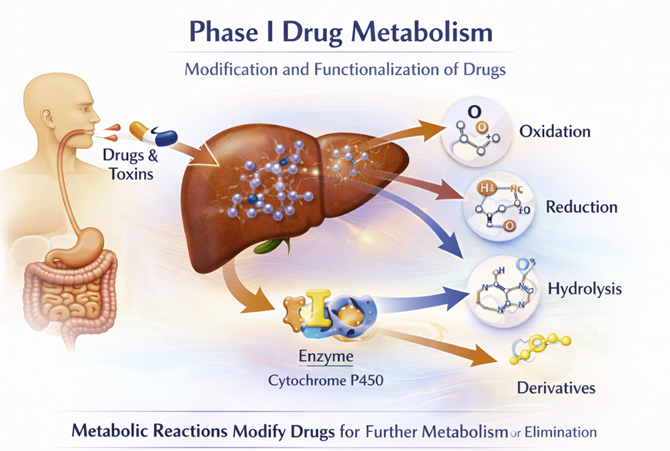
Drug metabolism is conventionally divided into two phases:
Phase I reactions introduce or expose a polar functional group on the drug molecule through oxidation, reduction, or hydrolysis.
The most important Phase I system is the hepatic cytochrome P450 (CYP) enzyme family which is a group of membrane-bound heme-containing enzymes located in the endoplasmic reticulum of hepatocytes.⁷
CYP enzymes belonging to the CYP1, CYP2, and CYP3 families are collectively responsible for approximately 70–80% of the metabolic biotransformation of drugs in clinical use.7
The most clinically significant isoforms are CYP3A4 (the most abundant hepatic CYP, responsible for ~50% of CYP-mediated metabolism), CYP2D6, CYP2C9, and CYP2C19.7
Phase I metabolism does not always inactivate a drug. It may:
Inactivate an active drug (most common outcome)
Activate a prodrug involving conversion of an inactive compound into its active form (e.g., codeine → morphine via CYP2D6; enalapril → enalaprilat)
Generate active metabolites with pharmacological properties similar to or different from the parent drug (e.g., diazepam → desmethyldiazepam)
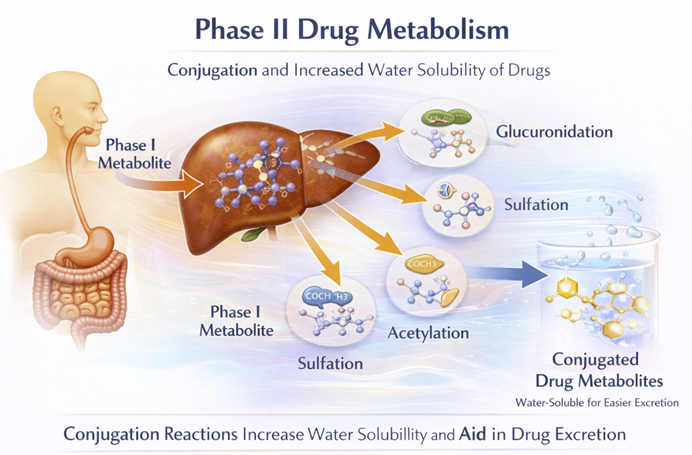
Phase II reactions conjugate the drug (or its Phase I metabolite) with an endogenous molecule such as glucuronic acid, sulfate, acetate, or glutathione, producing a highly water-soluble product that is pharmacologically inactive and readily excreted.8
Some drugs with appropriate polar groups bypass Phase I entirely and proceed directly to Phase II.
CYP Inhibition and Induction and Drug Interactions
The CYP enzyme system is not static. Its activity can be altered by drugs, foods, and other environmental factors, creating pharmacokinetic drug–drug interactions of major clinical importance:
CYP inhibition occurs when one drug blocks the enzymatic activity of a CYP isoform, reducing the metabolism of other drugs processed by the same enzyme.
The result is drug accumulation and potential toxicity.
Example: fluconazole inhibits CYP2C9, raising warfarin levels and dramatically increasing bleeding risk.7,9
CYP induction occurs when a drug upregulates CYP enzyme expression, accelerating the metabolism of co-administered drugs and potentially reducing them to subtherapeutic levels.
Example: rifampicin is one of the most potent CYP3A4 inducers known such that co-administration with many drugs (including oral contraceptives) can lead to treatment failure.7,9
Even non-drug substances matter.
Grapefruit juice irreversibly inhibits intestinal CYP3A4, increasing the bioavailability of statins, calcium channel blockers, and many other CYP3A4 substrates.
St. John's Wort induces CYP3A4, reducing plasma concentrations of HIV antiretrovirals, immunosuppressants, and anticoagulants.9
The CYP system also varies significantly between individuals due to genetic polymorphisms — a topic explored in depth in the Pharmacogenomics module (Module 5).
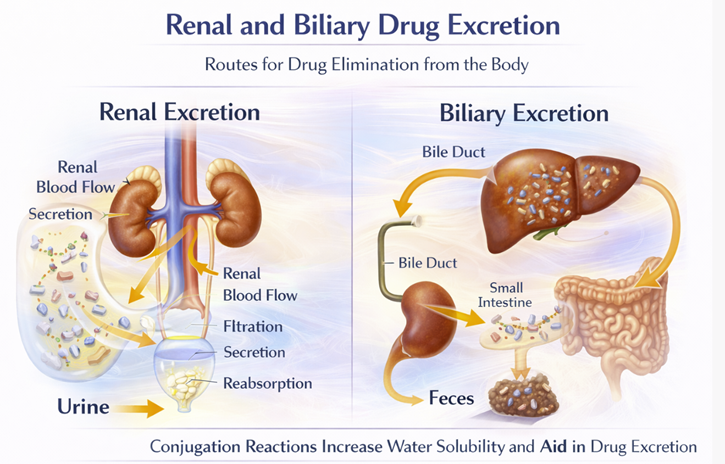
Excretion is the irreversible elimination of a drug or its metabolites from the body.
The kidneys are the primary organ of excretion for most drugs and their water-soluble metabolites; the biliary system (via the feces) is secondary.8
Renal excretion occurs through three mechanisms: glomerular filtration, active tubular secretion, and passive tubular reabsorption.
The net renal clearance of a drug reflects the balance of these three processes.
Lipophilic drugs are readily reabsorbed from the tubular lumen back into the circulation, which is why metabolism (to increase water solubility) is a prerequisite for efficient renal excretion.
Water-soluble drugs and metabolites are filtered and excreted with minimal reabsorption.
The clinical implication is direct: in patients with renal impairment, drugs that are primarily excreted unchanged by the kidneys accumulate.
Dose reduction or extended dosing intervals are required for renally cleared drugs in patients with reduced kidney function (e.g., aminoglycosides, digoxin, lithium, many antibiotics).
Failure to adjust for renal impairment is a leading cause of drug toxicity in hospitalized patients.
Biliary excretion is the main route for large molecular-weight drugs and their conjugated metabolites.
Some drugs excreted in bile are then reabsorbed from the intestine, a cycle called enterohepatic recirculation, which can dramatically prolong a drug's duration of action (e.g., some oral contraceptives, thyroid hormones, morphine).
Other minor excretion routes include the lungs (relevant for volatile anesthetics and alcohol), sweat, saliva, and breast milk.
The last is clinically important: drugs excreted in breast milk may expose breastfed infants to pharmacologically significant concentrations.
Pharmacokinetic and Pharmacodynamic Relationships
Pharmacokinetics does not stand alone. Rather pharmacokinetics and pharmacodynamics are linked.
This relationship gives rise to three clinically fundamental concepts:
The therapeutic window is the concentration range within which a drug produces the desired therapeutic effect without causing unacceptable toxicity.
Some drugs have wide therapeutic windows (most penicillins) such that modest overdosing is unlikely to result in harm.
Reaching steady state takes approximately 4–5 half-lives regardless of the dosing interval.
A drug's half-life (the time required for plasma concentration to fall by 50%) determines how frequently it must be dosed to maintain therapeutic concentrations.
These parameters are worked through quantitatively in the Pharmacokinetics chapter.
In the elderly, hepatic CYP450 activity decreases by ≥30% and renal function declines progressively, both increasing drug exposure.⁹
In neonates, hepatic enzyme systems are incompletely developed, and renal excretion is reduced, making this population exquisitely sensitive to drugs that adults clear efficiently.
In pregnancy, expanded plasma volume, altered protein binding, increased renal blood flow, and changes in CYP expression all alter drug disposition.
In patients with hepatic impairment, both Phase I and Phase II metabolism may be severely compromised, requiring major dose adjustments for hepatically cleared drugs.1,2
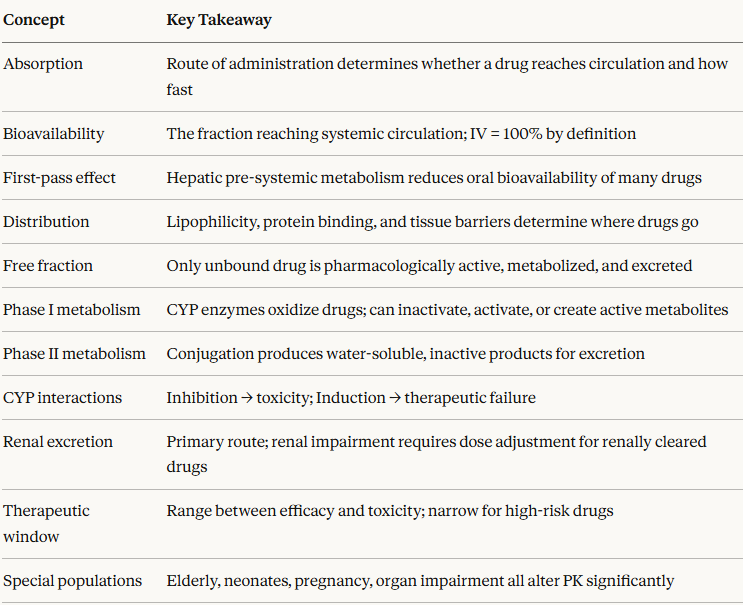
Module 2 Summary
|
|
|
|
|
|
|
|