Medical Pharmacology Chapter 1: General Principles: Overview and Introduction
|
|
|
|
|
|
|
-
Introduction to Pharmacology (Module 1)
-
-
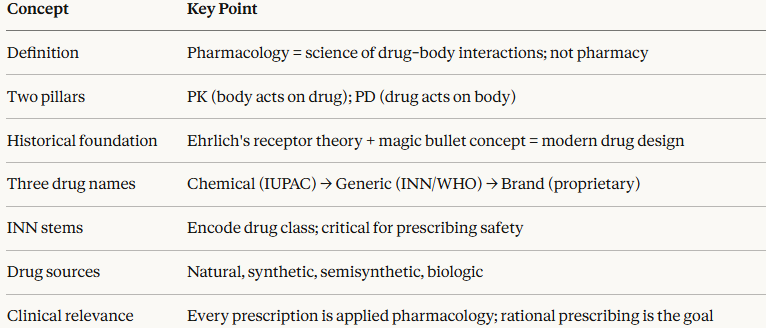
Pharmacology is the scientific discipline concerned with the interactions between chemical substances and living biological systems.
-
It encompasses the study of how drugs affect the body (pharmacodynamics) and how the body handles drugs (pharmacokinetics), together with their therapeutic applications and toxicological consequences.
-
Pharmacology is not pharmacy.
-
Pharmacy is a health services profession focused on dispensing, preparation, and clinical management of medicines.
-
-
Pharmacology is a research-based biomedical science whose findings underpin rational prescribing across every medical specialty.1
-
A drug is any chemical agent that alters biological processes.
-
A medicine is a drug that has been formally tested, regulated, and approved for use in the diagnosis, prevention, or treatment of disease.1
-
-
-
-
-
Pharmacokinetics (PK)
-
What the body does to the drug. The ADME processes (Absorption, Distribution, Metabolism, Excretion) determine how much drug reaches its target site and for how long.1,2
- Note: Detailed mathematical treatment of pharmacokinetic parameters (half-life, clearance, Vd, AUC) is covered in the dedicated Pharmacokinetics chapter on this site. This module provides the conceptual framework only.
-
-
Pharmacodynamics (PD)
-
What the drug does to the body. The molecular mechanisms by which a drug produces its effects, including receptor binding, signal transduction, and the resulting physiological changes.1,2
-
These two domains are deeply interdependent.
-
A drug with excellent PD potency is clinically worthless if poor PK prevents it from reaching therapeutic concentrations.
-
Conversely, favorable PK cannot compensate for a drug that lacks the right molecular interactions at its target site.
-
All subsequent modules return to this PK–PD framework as their organizing principle.
-
-
-
-
-
-
-
Brunton LL et al. Goodman & Gilman's. 13th ed. McGraw-Hill; 2017.
-
Katzung BG, Vanderah TW. Basic & Clinical Pharmacology. 15th ed. McGraw-Hill; 2021.
-
Easson LH, Stedman E. Studies on the relationship between chemical constitution and physiological action. Biochem J. 1933;27(4):1257–1266. Click for Article
-
Agranat I, Caner H, Caldwell J. Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov. 2002;1(10):753–768. Click for Article
-
Hutt AJ, Valentova J. The chiral switch: the development of single enantiomer drugs from racemates. Acta Facult Pharm Univ Comenianae. 2003;50:7–23. Click for reference
-
Calcaterra A, D'Acquarica I. The market of chiral drugs: chiral switches versus de novo enantiomerically pure compounds. J Pharm Biomed Anal. 2018;147:323–340. PMID 28865177. Click for Article
-
Smith SW. Chiral toxicology: it's the same thing — only different. Toxicol Sci. 2009;110(1):4–30. Click for Article
-
Why Molecular Shape Is the Fundamental Determinant of Drug Action
-
A drug molecule exerts its pharmacological effect by binding to a specific macromolecular target, most commonly a receptor protein, enzyme, ion channel, or transporter.
-
This binding is not a simple chemical collision; it is a precisely geometrically constrained interaction in which the three-dimensional shape of the drug must be complementary to the three-dimensional architecture of the binding site.
-
-
The receptor binding site is a chiral, three-dimensional cavity formed by the folding of a polypeptide chain.
-
Its walls are built from amino acid residues arranged in space, forming a surface of defined geometry, electrostatic character, and hydrogen-bond donor/acceptor topology.
-
A drug molecule that fits this cavity well, whose shape, charge distribution, and hydrogen-bonding groups are spatially complementary to those of the binding site, will bind with high affinity. A molecule that does not fit, even if chemically similar, will bind poorly or not at all.
-
-
Central Principle: Biological macromolecules such as receptors, enzymes, transporters, ion channels are themselves chiral.
-
They are constructed from L-amino acids, arranged in asymmetric three-dimensional structures.
-
A chiral binding site can distinguish between mirror-image ligands with the same precision that a left glove distinguishes a left hand from a right hand.
-
D/L System: A historical system based on structural relationship to D- and L-glyceraldehyde. Primarily used for amino acids and sugars. Not synonymous with the (+)/(−) optical rotation system, and a source of frequent confusion.
-
-
-
Chirality: Mirror Images That Cannot Be Superimposed
-
A molecule is chiral when it has a non-superimposable mirror image — when its mirror reflection is a distinct, non-identical molecule.
-
The most common structural basis for chirality in drug molecules is a stereocentre (also called a chiral centre or asymmetric carbon): a carbon atom bearing four different substituents, each pointing toward one corner of a tetrahedron.
-
-
Three nomenclature systems are used to describe chirality:
-
R/S System (CIP Rules): Based on priority ranking of substituents by atomic number. R (rectus) = clockwise arrangement of priorities; S (sinister) = counter-clockwise.
-
This describes absolute configuration and is the current standard in pharmacology and medicinal chemistry.
-
-
d/l or (+)/(−) System: Describes the optical rotation of plane-polarised light. (+) or d = dextrorotatory (rotates light clockwise); (−) or l = laevorotatory (rotates light counter-clockwise).
-
This is an observed experimental property that cannot be predicted from molecular structure alone and bears no fixed relationship to R/S designation.
-
-
D/L System: A historical system based on structural relationship to D- and L-glyceraldehyde. Primarily used for amino acids and sugars.
-
Not synonymous with the (+)/(−) optical rotation system, and a source of frequent confusion.
-
-
-
A pair of enantiomers have identical physical and chemical properties in an achiral environment — the same melting point, solubility, mass, and molecular formula.
-
They differ only in the direction they rotate plane-polarised light and in their interactions with chiral biological molecules, including all receptor binding sites.
-
-
-
The Three-Point Interaction Model
-
To understand why a chiral receptor can distinguish between enantiomers, Easson and Stedman (1933) proposed the three-point attachment model.
-
For a drug to achieve stereospecific binding, at least three points on the drug molecule must interact simultaneously with three complementary points on the receptor.
-
-
If the drug is chiral and binding requires three simultaneous contacts, one enantiomer will achieve all three contacts with appropriate geometry while the other can achieve at most two with the third contact projecting in the wrong spatial direction.
-
The result is a dramatic difference in binding affinity and therefore pharmacological potency between the two mirror images.
-
-
This model has been refined over decades but its core insight remains valid.
-
It is not enough to have the right functional groups; they must be in the right spatial arrangement relative to each other and to the binding site.
-
Shape and geometry are inseparable from pharmacological activity.
-
-
-
Enantiomers: Four Patterns of Pharmacological Difference
-
When a drug has a stereocentre, the two enantiomers can differ in pharmacology in four clinically important patterns.
-
Pattern 1 — One Active, One Inert: One enantiomer carries the therapeutic activity; the other is pharmacologically inert at the target receptor.
-
The inert enantiomer is simply carried along as pharmacological dead weight in a racemic formulation.
-
Examples: S-ketamine (the active anaesthetic/antidepressant enantiomer); L-DOPA (the only form crossing the blood-brain barrier and entering the dopaminergic pathway).
-
-
Pattern 2 — Different Potencies: Both enantiomers bind the target receptor but with different affinities.
-
One is more potent; the other has the same qualitative activity but requires a higher concentration for equivalent effect.
-
Example: S-warfarin is approximately five times more potent as an anticoagulant than R-warfarin, though both inhibit vitamin K epoxide reductase.
-
-
Pattern 3 — Different Qualitative Pharmacological Actions: Each enantiomer exerts qualitatively different pharmacological effects, binding different receptors or targets, producing distinct and sometimes opposing actions in the same patient.
-
Examples: the enantiomers of sotalol differ in β-blocking and class III antiarrhythmic activity; quinine and quinidine are diastereomers with entirely different clinical applications (antimalarial vs antiarrhythmic).
-
-
Pattern 4 — One Enantiomer Causes Toxicity: The two enantiomers have different safety profiles, one produces the desired therapeutic effect while the other is responsible for adverse effects or toxicity.
-
The defining example is thalidomide, where R-(+)-thalidomide produces sedation and S-(−)-thalidomide is responsible for teratogenesis through binding to cereblon and disrupting limb bud development.
-
-
Clinically Notable Chiral Drug Pairs 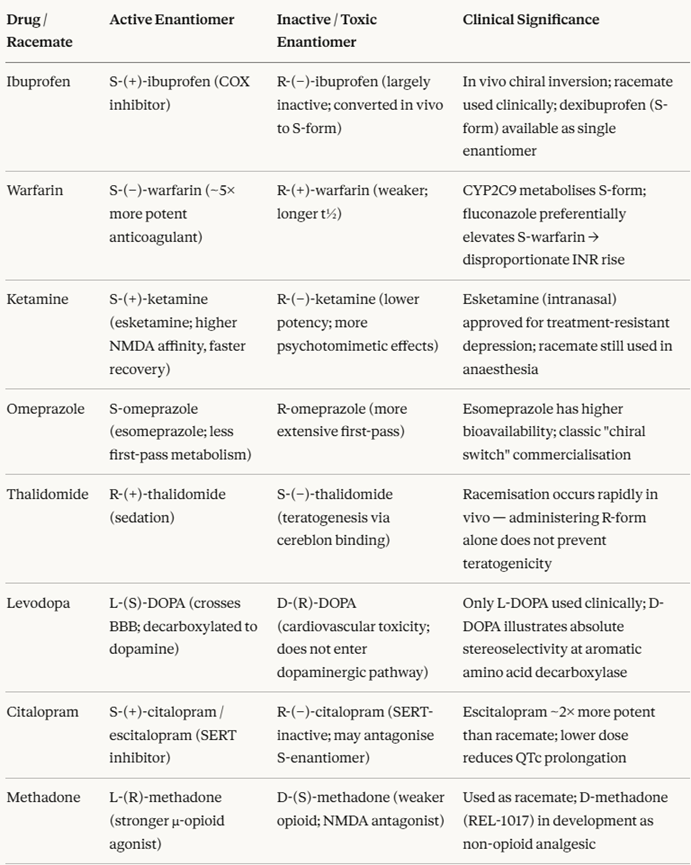
-
-
-
Racemic Mixtures, Chiral Switches, and Drug Development
-
Historically, the majority of chiral drugs were developed and marketed as racemates, equimolar mixtures of both enantiomers, because separating enantiomers on a large scale was technically difficult and expensive.
-
This meant that patients routinely received a pharmacological mixture: one enantiomer exerting the desired therapeutic effect, the other contributing to adverse effects, or simply not contributing to efficacy at all.
-
-
The chiral switch is the development of a single-enantiomer drug from a previously racemic agent.
-
The rationale is pharmacologically sound: eliminating the inactive or toxic enantiomer should improve the efficacy-to-toxicity ratio, simplify pharmacokinetics, and reduce the total drug burden on the patient.
-
Regulatory agencies (FDA, EMA) now require full stereochemical characterisation of new chiral drug candidates and expect justification for the choice of racemate over single enantiomer.
-
-
Levalbuterol (R-albuterol) illustrates this in respiratory medicine.
-
Racemic albuterol (salbutamol) consists of R- and S-albuterol.
-
Bronchodilatory action resides almost entirely in the R-(−)-enantiomer, which activates β₂-adrenoceptors.
-
The S-(+)-enantiomer has negligible bronchodilatory effect and may at high concentrations promote airway inflammation and hyperresponsiveness.
-
Levalbuterol is available as a single-enantiomer inhaler at half the total drug load, producing equivalent bronchodilation, though whether this translates to a clinically meaningful advantage over the racemate in routine practice remains to be resolved.
-
-
-
A critical complication is chiral inversion which is enzymatic conversion of one enantiomer to the other after administration.
-
Ibuprofen is the paradigm: the inactive R-enantiomer is partially converted to the active S-form in the liver by 2-arylpropionyl-CoA epimerase.
-
This means that predicting the clinical behaviour of a chiral drug from in vitro data alone is insufficient given that the in vivo fate of each enantiomer must be independently characterized.
-
-
-
-
Pharmacokinetic Stereoselectivity
-
Stereoselectivity is not confined to receptor interactions.
-
Every major pharmacokinetic process can be stereoselective, because each involves chiral proteins as molecular machinery.
-
-
Absorption: Intestinal drug transporters (P-glycoprotein, PEPT1, OCT) are stereoselective.
-
L-amino acids are actively transported while D-amino acids are not.
-
Drug enantiomers that are substrates for these carriers may differ substantially in bioavailability.
-
-
Protein Binding: Plasma proteins, albumin and α₁-acid glycoprotein are chiral and bind enantiomers with different affinities.
-
The S-enantiomer of propranolol binds α₁-AGP more tightly than the R-enantiomer, producing different free fractions, volumes of distribution, and hepatic extraction ratios between the two forms.
-
-
Metabolism: CYP450 enzymes have chiral active sites and metabolise enantiomers at different rates and by different pathways.
-
S-warfarin is metabolised primarily by CYP2C9; R-warfarin by CYP1A2 and CYP3A4.
-
Drug interactions that inhibit CYP2C9 (such as fluconazole, amiodarone, or miconazole) therefore affect the enantiomers asymmetrically selectively elevating the more potent S-warfarin and producing disproportionate increases in anticoagulant effect that are not predicted by total warfarin concentration alone.
-
-
The clinical consequence:
-
When a racemic drug is administered, the plasma concentration ratio of R to S enantiomers at steady state is often not 1:1.
-
It is determined by the relative rates of absorption, distribution, metabolism, and excretion of each enantiomer independently.
-
The clinical response therefore reflects this emergent steady-state mixture, not the administered ratio.
-
-
-
-
-
Diastereomers and Multiple Stereocenters
-
When a drug molecule contains two or more stereocentres, the stereochemical complexity increases substantially.
-
With n stereocentres, up to 2ⁿ stereoisomers are possible. Isomers that differ at some but not all stereocentres are called diastereomers
-
They are not mirror images of each other, and they have different physical and chemical properties (different melting points, solubilities, partition coefficients, and receptor affinities).
-
-
-
Ephedrine and pseudoephedrine are diastereomers, differing at one of their two stereocentres.
-
They have markedly different pharmacological profiles: ephedrine is a mixed direct/indirect sympathomimetic with significant cardiovascular activity; pseudoephedrine is primarily a nasal decongestant with much less cardiac effect.
-
Their different properties arise from the different three-dimensional orientation of the same functional groups relative to each other.
-
-
Quinine and quinidine are diastereomers with completely different clinical applications:
-
Quinine is an antimalarial and quinidine is a class Ia antiarrhythmic.
-
This is perhaps the most striking example of how a single stereocentre difference can produce drugs with entirely distinct indications, mechanisms, and adverse effect profiles.
-
-
-
-
Chirality Summary Table 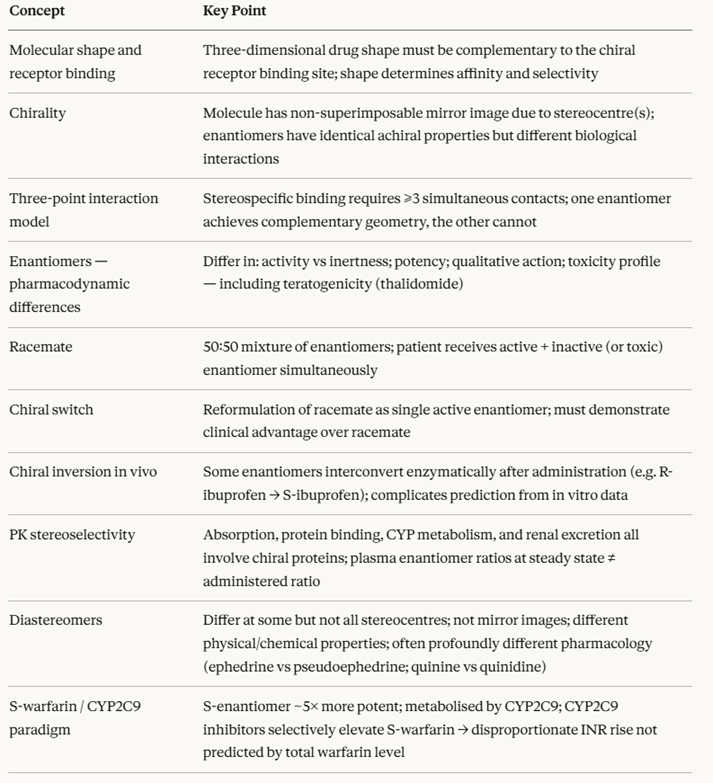
-
-
-
A Brief History: From Empiricism to Molecular Pharmacology
-
Ancient pharmacology relied entirely on empirical observation e.g. willow bark (salicylates), opium poppies, foxglove (digitalis) without any mechanistic understanding.
-
The first Western pharmacological catalogue was compiled by Dioscorides in the 1st century AD.10
-
The 19th century transformation was driven by chemical isolation of active ingredients: morphine, strychnine, atropine, quinine, digitalis glycosides.
-
Oswald Schmiedeberg (1838–1921) formally established pharmacology as a scientific discipline, wrote the first pharmacology textbook, and trained the generation who founded departments worldwide.10
-
-
Paul Ehrlich (1854–1915) made two foundational contributions.
-
First, his receptor theory proposed that cells possess specific surface structures capable of binding selectively with extracellular molecules, what we today call receptors and ligands.4
-
Second, his concept of the Zauberkugel (magic bullet), a drug selectively targeting disease-causing agents without harming the host, was validated by the discovery of arsphenamine (Salvarsan, 1909), the first synthetic treatment for syphilis.
-
This principle underlies all targeted drug design, including modern monoclonal antibodies and tyrosine kinase inhibitors.4,5
-
-
-
Raymond Ahlquist's 1948 proposal of α- and β-adrenergic receptor subtypes led Sir James Black to develop propranolol (1965), the first clinically useful receptor-blocking drug, setting the template for translating receptor pharmacology directly into clinical therapeutics.3
-
-
Contemporary pharmacology has seen a transition driven by large-molecule biologics, monoclonal antibodies, chimeric proteins, gene therapies, representing a fundamental departure from the classical small-molecule pharmacology paradigm.
-
Biologics are produced by living cells, may be immunogenic, and many display nonlinear dose–response relationships.3
-
-
-
Drug Nomenclature: The Three-Name System
Every drug in clinical use carries at least three distinct names, each serving a different purpose.
Confusion among these names is a common source of medication errors and prescribing mistakes; understanding the nomenclature system is therefore a clinical safety competency, not merely an academic one.
The chemical name describes the precise molecular structure of the drug according to rules set by the International Union of Pure and Applied Chemistry (IUPAC).
Chemical names are scientifically unambiguous but are typically far too complex for clinical use.
For example, the IUPAC name for aspirin is 2-(acetyloxy)benzoic acid.
Chemical names are used primarily in drug discovery, patent filings, and regulatory chemistry documentation.7
The Generic (Nonproprietary) Name
The generic name, technically called the International Nonproprietary Name (INN) when assigned by the World Health Organization, is the universally standardized name for a drug's active ingredient.
The INN system was formally established in 1953 when the World Health Assembly passed resolution WHA3.11, mandating an expert WHO committee to select non-proprietary names for drugs for use in pharmacopoeias, prescribing, and scientific literature worldwide.8
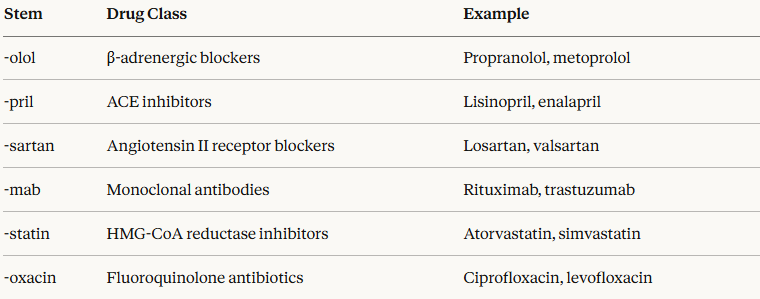
A key aspect of the INN system lies in its use of systematic stems, short syllabic fragments embedded in a drug's name that indicate its pharmacological class.
This approach allows clinicians and pharmacists to infer pharmacological properties from the name itself:
INNs are placed in the public domain by the WHO and may be used without restriction by any party.7
WHO policy prohibits commercial trademarks from being derived from INNs or from incorporating their stems, as this would compromise patient safety by introducing confusion into the nomenclature system.7
The brand name is the manufacturer's trademarked commercial identifier, used for marketing purposes.
A single generic drug may be sold under multiple brand names by different manufacturers in different countries.
Brand names are approved by national regulatory authorities (e.g., the FDA in the United States) to ensure they are sufficiently distinct from other existing drug names.
This three-name system is of significant clinical importance.
A prescriber who knows only brand names is dependent on the originator manufacturer and may not recognize that an unfamiliar generic drug is pharmacologically identical to one they know well.
Conversely, a clinician who understands INN stems can rapidly decode the drug class and anticipated pharmacological properties of an unfamiliar compound.
Sources and Classification of Drugs
Drugs originate from several distinct sources.
Natural sources include plants (morphine from Papaver somniferum, digoxin from Digitalis lanata, taxol from the Pacific yew), animals (insulin was originally derived from bovine and porcine pancreas), and microorganisms (penicillin from Penicillium notatum, statins originally derived from fungal metabolites).1,2
Opium Poppy
Attribution: tanja niggendijker from apeldoorn, the netherlands, CC BY 2.0 <https://creativecommons.org/licenses/by/2.0>, via Wikimedia Commons
https://commons.wikimedia.org/wiki/File:Papaver_somniferum_flowers.jpg
Digitalis lanata
Attribution: User: Haplochromis, CC BY-SA 3.0 <http://creativecommons.org/licenses/by-sa/3.0/>, via Wikimedia Commons
https://commons.wikimedia.org/wiki/File:Digitalis_lanata_1.JPG
Synthetic compounds are produced by organic chemical synthesis.
Most modern small-molecule drugs are synthetic.
The structure-activity relationship (SAR), the systematic study of how changes in molecular structure alter pharmacological activity, emerged as a discipline in the early 20th century and remains central to drug development.10
Semisynthetic compounds are natural products that have been chemically modified to improve their pharmacological profiles.
Many antibiotics such as ampicillin, amoxicillin are semisynthetic derivatives of naturally occurring penicillin.1
Biologics are large-molecule drugs produced by biological systems (bacteria, mammalian cells, yeast).
This category includes monoclonal antibodies, recombinant proteins, vaccines, and gene therapies.
Biologics represent a fundamental departure from classical small-molecule pharmacology in their production, mechanism, dose-response characteristics, and immunogenicity.3
Classification systems organize drugs to facilitate learning, prescribing, and oversight of pharmacological actions in clinical settings.
The most comprehensive international system is the Anatomical Therapeutic Chemical (ATC) Classification System, maintained by the WHO.
The ATC system organizes drugs into five hierarchical levels based on the organ system they act on, their therapeutic use, and their chemical properties.2
Pharmacology courses typically organize content by drug class (e.g., beta-blockers, ACE inhibitors) or by organ system (cardiovascular, CNS, respiratory), both of which map onto the ATC framework.
Clinical Pharmacology: Translating Science to Clinical Practice
Clinical pharmacology occupies the interface between the basic science of pharmacology and the care of individual patients.
WHO's definition of clinical pharmacoology:
clinical pharmacologists:
Work to improve patient care by promoting the safe and more effective use of drugs
Undertake research on drug actions in humans
Provide objective, evidence-based therapeutic information to ethics, regulatory, and pricing bodies
Contribute to medicines policy for governments.6
For medical students, the practical implication is this: every prescribing decision is applied pharmacology.
The prescriber selects a drug (implicitly choosing a mechanism of action), selects a dose and route (implicitly making pharmacokinetic assumptions), and monitors for both therapeutic and adverse effects (implicitly testing the drug's pharmacodynamic profile in an individual patient).
Pharmacology is not a subject to be learned and forgotten after the preclinical years because pharmacology is the scientific foundation of every clinical encounter involving medication.
The concept of rational prescribing, emphasized by both Goodman & Gilman and Katzung, means selecting drugs based on an understanding of their mechanism, their pharmacokinetic properties, and their evidence base — rather than habit, marketing influence, or familiarity1,2
This framework is progressively being refined by pharmacogenomics (covered in a later module), which promises to individualize prescribing based on a patient's genetic profile.



|
|
|
|
|
|
|
|