Medical Pharmacology Chapter 1: General Principles: Overview and Introduction
|
|
|
|
|
|
|
-
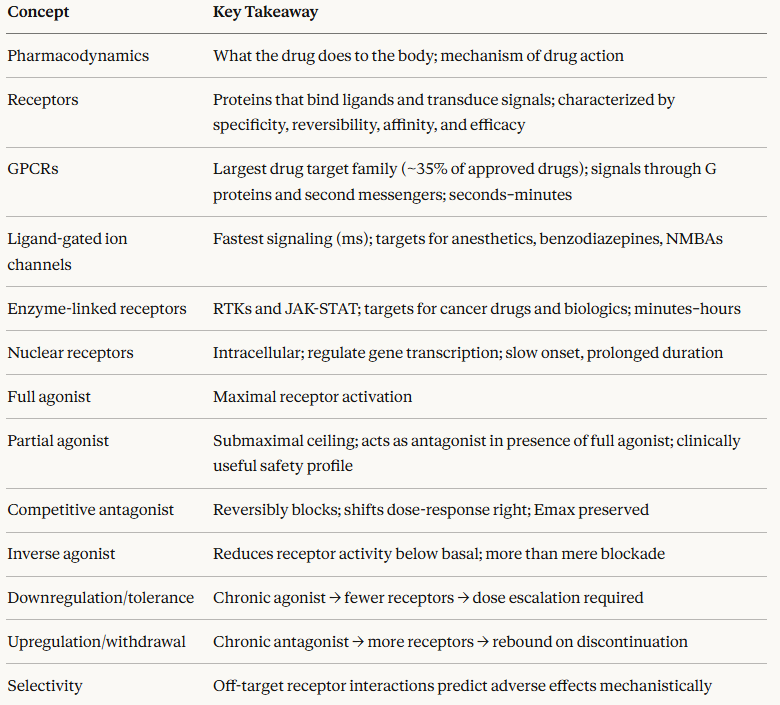
Pharmacodynamics (PD) is the study of the biochemical and physiological effects of drugs on the body, and the mechanisms by which those effects are produced.
-
If pharmacokinetics answers the question "What does the body do to the drug?", pharmacodynamics answers the complementary question "What does the drug do to the body?"1
-
The pharmacodynamic relationship is concentration-dependent: the effect of a drug at its target site changes as the concentration changes, producing the dose–response relationship that underlies all of clinical prescribing.
-
Understanding pharmacodynamics means understanding not just that a drug works, but how it works at the molecular level and why that mechanism predicts both its therapeutic effects and its adverse effects.1,2
-
-
 Most drugs
exert their effects by interacting with one of a defined set of
molecular targets.
Most drugs
exert their effects by interacting with one of a defined set of
molecular targets. -
These include receptors (the primary focus of this module), enzymes, ion channels, and transporter proteins.
-
Each target class has a distinct mechanism of action, a characteristic time course of effect, and a specific set of clinical implications.
-
-
-
-
A
receptor is a protein molecule that is located on the
cell surface, within the cell membrane, or intracellularly and
whose function is to recognize and bind specific ligands
(endogenous signaling molecules or exogenous drugs) and to
transduce that binding event into a cellular response.9
-
Principles of receptor pharmacology:
-
-
Receptors evolved to respond to specific endogenous molecules such as neurotransmitters, hormones, growth factors, and lipid mediators.
-
Drugs take advantage of this specificity by mimicking or blocking the effects of natural ligands.
-
Cclinical effects of drugs are largely a reflection of which receptors it binds and with what affinity and efficacy.1
-
-
-
-
Drug–receptor binding is usually reversible, forming a dynamic equilibrium between bound and unbound states.
-
Reversibility is what allows a drug's effect to be modulated and terminated.
-
Irreversible binding (covalent or quasi-irreversible) is less common but clinically important.
-
For example, aspirin irreversibly acetylates cyclooxygenase enzymes, which is why its antiplatelet effect outlasts its plasma half-life, persisting for the full lifespan of the platelet (7–10 days).1
-
-
-
-
-
Affinity refers to the strength of the drug–receptor binding interaction (how tightly a drug binds).
-
A drug with high affinity binds at very low concentrations.
-
Affinity is quantified by the dissociation constant (Kd): the lower the Kd, the higher the affinity.
-
Detailed treatment of affinity and Kd is in the Pharmacodynamics chapter.
-
-
-
-
-
-
Efficacy (or intrinsic activity) refers to the ability of a drug–receptor complex to produce a biological response once bound.
-
A drug can have high affinity for a receptor but zero efficacy.
-
This interaction describes an antagonist, which occupies the receptor without activating it.1,2
-
-
-
-
-
The Four Receptor Superfamilies
-
Receptors are organized into four major superfamilies based on their structure, location, and signaling mechanism.
-
This classification maps directly onto clinical pharmacology, because the receptor type determines the onset, duration, and nature of a drug's effect.1,2,3
-
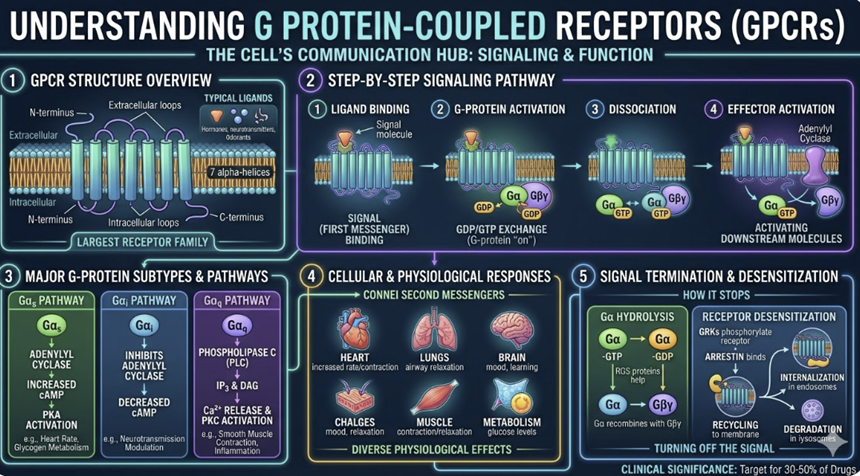
G Protein-Coupled Receptors (GPCRs)
-
GPCRs are the largest family of drug targets in medicine.
-
Approximately 35% of all FDA-approved drugs act through GPCRs, making them the single most important receptor class in clinical pharmacology.4
-
Structurally, GPCRs are seven-pass transmembrane proteins since the receptor spans the cell membrane seven times with an extracellular ligand-binding domain and an intracellular domain coupled to a heterotrimeric G protein (composed of α, β, and γ subunits).
-
When a
ligand binds, the G protein is activated, dissociating into its
α and βγ subunits, each of which can activate downstream
effectors.4
-
-
-
The nature of the downstream effects depend on which G protein subtype is coupled:
-
-
Clinically important GPCR families include adrenergic receptors (targets of α-blockers, α-agonists), muscarinic receptors (targets of anticholinergics), opioid receptors (targets of opioid analgesics), and dopamine receptors (targets of antipsychotics).
-
The breadth of this list emphasizes the central role GPCRs play in regulating virtually every major physiological system.4
-
Signaling speed: GPCRs produce effects within seconds to minutes through second messenger cascades.
-
-
-
-
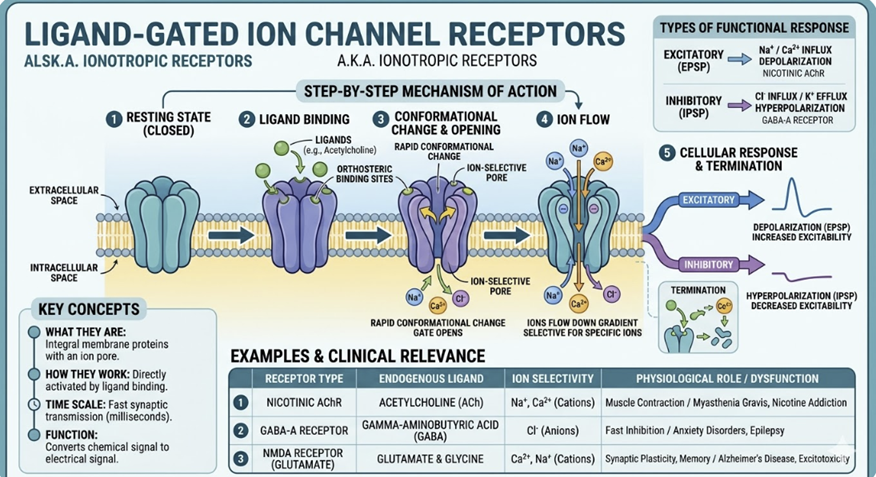
Ligand-Gated Ion Channels (LGICs)
-
-
Ligand-gated ion channels are transmembrane protein complexes that open to allow specific ions to flow across the cell membrane when a ligand binds.
-
Because ion flux changes membrane potential almost instantaneously, ligand-gated ion channels mediate the fastest drug effects in pharmacology on the order of milliseconds.3
-
-
Important examples include:
-
Nicotinic acetylcholine receptors (nAChR)
-
Cation channels (Na⁺/K⁺) at the neuromuscular junction and autonomic ganglia; target of neuromuscular blocking agents (e.g., succinylcholine, rocuronium)
-
-
Glutamate receptors (NMDA, AMPA)
-
Cation channels mediating excitatory neurotransmission; target of ketamine (NMDA antagonist)3
-
-
GABA-A receptors
-
Chloride channels; the primary inhibitory neurotransmitter receptor in the CNS.
-
These receptors target of benzodiazepines (which potentiate GABA-induced Cl⁻ influx and membrane hyperpolarization) and general anesthetics.
-
-
-
-
-
-
Enzyme-Linked Receptors (Receptor Tyrosine Kinases and Related)
-
Enzyme-linked receptors are transmembrane proteins that either possess intrinsic enzymatic activity or are closely associated with intracellular enzymes.
-
The most important subclass is the receptor tyrosine kinases (RTKs), which phosphorylate tyrosine residues on intracellular signaling proteins following ligand binding.4
-
RTKs are the targets of a rapidly expanding class of cancer drugs, the tyrosine kinase inhibitors (e.g., imatinib targeting BCR-ABL, gefitinib targeting EGFR, trastuzumab targeting HER2).
-
These drugs represent one of the most direct translations of receptor pharmacology into targeted cancer therapy, embodying Paul Ehrlich's original "magic bullet" concept at the molecular level.1
-
-
JAK-STAT receptors are a related class that lack intrinsic kinase activity but recruit and activate cytoplasmic JAK kinases upon ligand binding.
-
These are targets of JAK inhibitors now widely used in rheumatoid arthritis and inflammatory bowel disease (e.g., tofacitinib, baricitinib).3
-
Signaling speed: Minutes to hours, as effects require activation of kinase cascades.
-
-
-
-
-
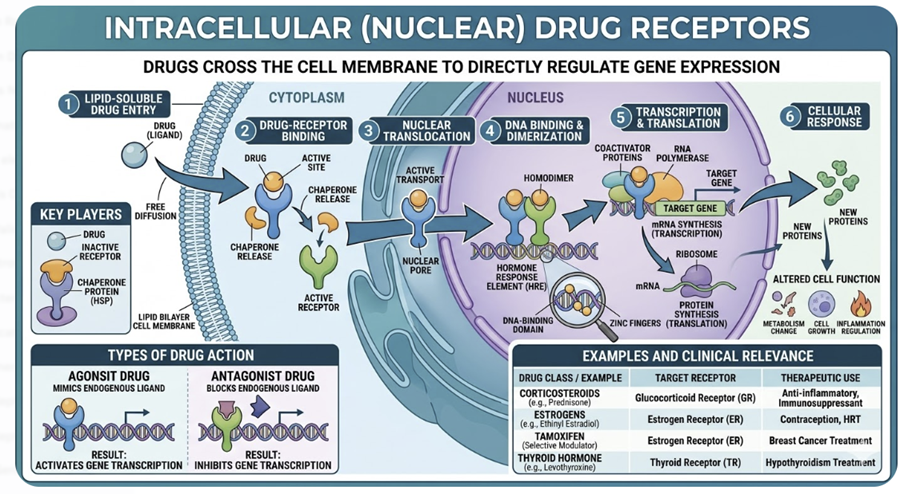
Intracellular (Nuclear) Receptors
-
-
Nuclear receptors are located inside the cell — in the cytoplasm or nucleus — and require the ligand to cross the cell membrane to reach them.
-
Consequently, their ligands are typically lipophilic small molecules: steroid hormones (glucocorticoids, estrogens, androgens), thyroid hormones, vitamin D, and retinoids.1
-
-
When a lipophilic ligand diffuses into the cell and binds its receptor, the resulting complex translocates to the nucleus (if not already there), binds to specific DNA regulatory sequences called hormone response elements (HREs), and directly regulates gene transcription.
-
This activity results in new protein synthesis, representing a different mechanism from the rapid signaling of GPCRs or ion channels.1,2
-
This mechanism explains several clinically important features of glucocorticoids and other steroid drugs:
-
Slow onset: Effects require hours to days because new protein synthesis takes time.
-
Prolonged duration: Effects outlast the drug's plasma half-life because newly synthesized proteins persist.
-
Broad systemic effects: Transcriptional regulation of hundreds of genes produces wide-ranging physiological effects.
-
Withdrawal syndromes: Abrupt discontinuation of exogenous glucocorticoids can precipitate adrenal crisis because the HPA axis has been suppressed representing a direct pharmacodynamic consequence of receptor-mediated transcriptional suppression.
-
-
Signaling speed: Hours to days corresponding to the slowest of all receptor classes.
-
-
-
-
-
-
Not all drugs act through traditional receptors.
-
Three
other important molecular target classes deserve
conceptual recognition.1,2
-
Enzymes are inhibited by many major drug classes.
-
Aspirin inhibits COX enzymes
-
ACE inhibitors block angiotensin-converting enzyme
-
Statins inhibit HMG-CoA reductase
-
Many antibiotics inhibit bacterial cell wall synthesis enzymes
-
Proton pump inhibitors irreversibly block the H⁺/K⁺-ATPase.
-
Enzyme inhibition is generally well-suited to small-molecule drugs because enzyme active sites are geometrically defined binding pockets.
-
-
-
Ion channels can be targeted by drugs that act directly on the channel protein (distinct from the LGIC mechanism above).
-
Voltage-gated sodium channels are blocked by local anesthetics (lidocaine, bupivacaine) and antiepileptics (phenytoin, carbamazepine).
-
Voltage-gated calcium channels are blocked by calcium channel blockers (amlodipine, verapamil).
-
These drugs bind to the channel protein and alter its conductance or gating properties, independent of a separate receptor binding event.
-
-
-
Transporter proteins are the targets of several important drug classes.
-
Selective serotonin reuptake inhibitors (SSRIs) block the serotonin transporter (SERT), increasing synaptic serotonin concentration.
-
Sodium-glucose cotransporter-2 (SGLT2) inhibitors block renal glucose reabsorption, lowering blood glucose in type 2 diabetes.
-
Metformin acts partly by inhibiting mitochondrial respiratory chain transporters.
-
Targeting transporters allows drugs to alter the concentration of endogenous molecules at physiologically important sites without directly binding a receptor.
-
-
-
-
-
-
-
After a drug binds its receptor, the nature of the interaction determines the pharmacological consequence.
-
Vocabulary of ligand classification is essential for understanding drug action and predicting clinical effects.5
-
-
Full agonists bind to a receptor and produce the maximal biological response that the receptor-effector system is capable of generating.
-
The full agonist exhibit both high affinity with respect to receptor binding and high intrinsic efficacy as the drug maximally activates the receptor.
-
Clinical examples include morphine (full agonist at μ-opioid receptors) and adrenaline (full agonist at adrenergic receptors).
-
-
-
Partial agonists bind to a receptor and activate it, but even if the drug occupies all of the available target receptors, a submaximal response response occurs, indicative of a lower ceiling effect compared to a full agonist.
-
This result is due to the partial agonists having a lower intrinsic efficacy despite similar (or even higher) affinity.
-
If a partial agonist is present alongside a full agonist competing for the same receptor, the partial agonist reduces the full agonist effect by competing with the full agonist for receptor occupancy.
-
-
Partial agonism has direct clinical utility:
-
Buprenorphine is a partial agonist at the μ-opioid receptor, providing meaningful analgesia and reducing cravings in opioid use disorder, while its ceiling effect limits the risk of respiratory depression.
-
This property makes the partial agonist, buprenorphine inherently safer than full opioid agonists for maintenance therapy.
-
-
Aripiprazole is a partial agonist at dopamine D2 receptors, providing antipsychotic efficacy while avoiding the full dopamine blockade responsible for the extrapyramidal side effects of older antipsychotics.
-
Varenicline is a partial agonist at nicotinic acetylcholine receptors, reducing nicotine withdrawal symptoms while blocking the reinforcing effects of smoked nicotine5,6
-
-
Antagonists bind to receptors with affinity but without intrinsic efficacy in that antagonist occupy the receptor without activating it.
-
Their clinical effect is based on preventing endogenous ligands or exogenous agonists from binding, thereby blocking the receptor's physiological response.
-
The antagonist may compete with the agonist for binding sites.
-
Another possibility is that the antagonist might bind effectively "irreversibly" to the receptor, making binding by an agonist unlikely.
-
-
-
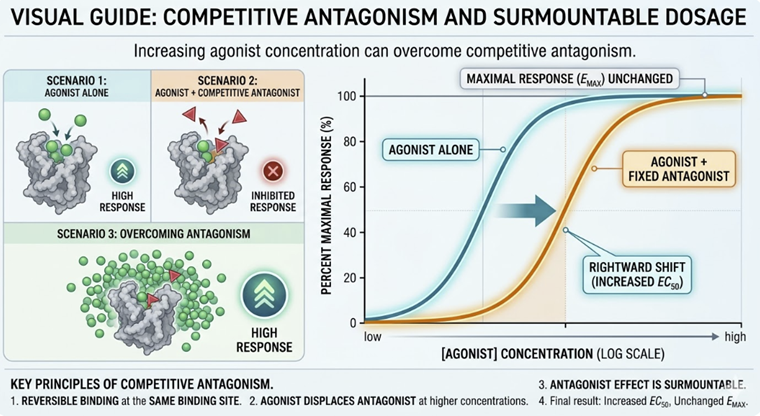
Competitive antagonists compete reversibly with agonists for the same binding site.
-
Their effect can be overcome by increasing the agonist concentration.
-
They shift the dose–response curve to the right without reducing the maximum effect (Emax).
-
-
Examples: naloxone (opioid antagonist), propranolol (β-adrenergic antagonist), atropine (muscarinic antagonist).
-
-
-
-
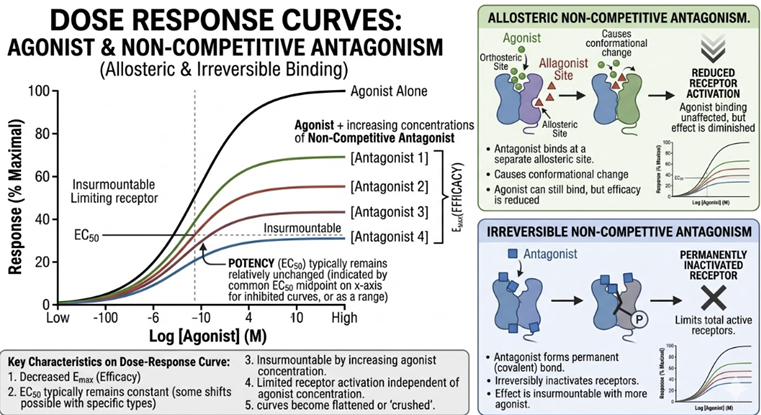
Non-competitive antagonists bind to a site distinct from the agonist binding site (allosteric) or bind irreversibly, preventing receptor activation regardless of agonist concentration.
-
Note reduction in Emax.
-
-
Example: phenoxybenzamine (irreversible α-adrenergic blocker).5
-
-
-
-
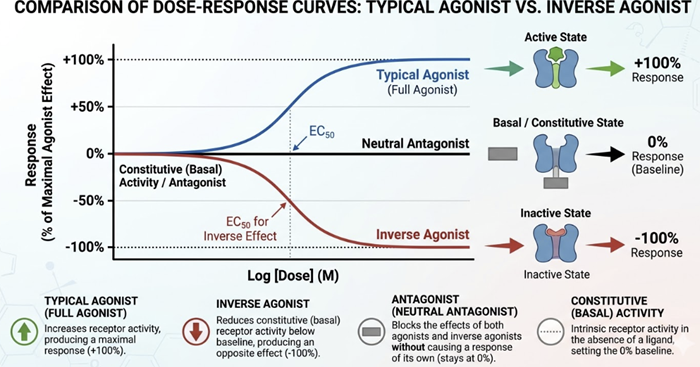
Inverse agonists are a conceptually important and increasingly recognized drug category.
-
Most receptors exist in equilibrium between active and inactive conformational states, even without any ligand bound, described as constitutive receptor activity.
-
Conventional antagonists are truly neutral: they occupy the receptor without shifting this equilibrium.
-
Inverse agonists, by contrast, bind preferentially to the inactive conformation of the receptor, thereby reducing activity below basal levels.6
-
-
-
This distinction is clinically relevant given that most drugs previously classified simply as "antihistamines" (H1 blockers) are now recognized as inverse agonists rather than neutral antagonists.
-
Their ability to reduce constitutive H1 receptor activity.not merely block histamine which may contribute to their clinical potency.6
-
-
-
-
-

-
Receptors are not fixed targets.
-
Prolonged exposure to drugs, particularly agonists and antagonists induce adaptive regulatory changes in receptor number and sensitivity which affect clinical responses.7,8
-
Desensitization is a rapid, short-term reduction in receptor responsiveness following sustained agonist exposure, typically occurring over minutes.
-
Desensitization often deptends on phosphorylation of intracellular receptor domains that uncouple the receptor from its G protein, reducing signaling efficiency without changing receptor number.
-
Desensitization may be thought of as a protective homeostatic mechanism.7
-
-
-
Downregulation is a slower, more sustained reduction in the number of functional receptors following prolonged agonist exposure, occurring over hours to days through receptor internalization and degradation.
-
Downregulation explains pharmacodynamic tolerance: the progressive decrease in drug effect that occurs with repeated administration, requiring escalating doses to maintain the same response.
-
Opioid tolerance that develops with chronic opioid use occurs in part by μ-opioid receptor downregulation mediated by arrestin-dependent endocytosis.7
-
-
Upregulation is the reciprocal phenomenon to downregulation in that here chronic receptor blockade (by an antagonist) removes normal inhibitory signals and triggers compensatory increases in receptor number and sensitivity.
-
This mechanism underlies withdrawal syndromes described as an exaggerated physiological responses that follow abrupt discontinuation of long-term antagonist therapy:
-
-
Chronic beta-adrenergic blockade upregulates β-receptor density.
-
Abrupt discontinuation exposes these supranumerary receptors to circulating catecholamines, potentially precipitating severe rebound tachycardia, angina, or hypertensive crisis which is especially dangerous in patients with coronary artery disease.8
-
-
-
-
Chronic clonidine administration, an α2-adrenergic agonist that downregulates central sympathetic tone, results in compensatory upregulation of sympathetic outflow pathways.
-
Abrupt discontinuation can cause a hypertensive rebound crisis.8
-
-
-
-
-
Pharmacodynamics and Drug Target Selectivity
-
A recurring theme in pharmacodynamics is the concept of selectivity which descibes the degree to which a drug preferentially interacts with one receptor subtype or molecular target over others.
-
No drug is perfectly selective; all drugs produce off-target effects to varying degrees, and many adverse drug reactions are simply the pharmacological consequence of interacting with unintended but predictable secondary targets.1,2
-
-
Understanding selectivity allows adverse effects to be viewed mechanistically.
-
A drug that blocks histamine H1 receptors will cause sedation if it also crosses the blood-brain barrier to block central H1 receptors.
-
A non-selective β-blocker will cause bronchoconstriction by blocking β₂ receptors in airway smooth muscle.
-
An antipsychotic that blocks both dopamine D2 receptors and muscarinic M1 receptors will produce both antipsychotic effects and anticholinergic side effects.
-
Adverse effects follow from considering the basis of drug selectivity.
-
-
|
|
|
|
|
|
|
|