|
|
|
|
|
|
|
Chapter 3: General Principles: Pharmacodynamics
|
|
|
|
|
|
|
Audio Overview: Pharmacodynamics: Drug Selective, Specificity and the Therapeutic Index (Extended)
Audio Overview: Drug Selective, Specificity and the Therapeutic Index (Brief)
Every drug that has ever caused an adverse effect did so for one of two reasons:
Ether it reached an unintended molecular target, or
It reached its intended target in an unintended tissue or at an unintended time.
Both of these are failures of selectivity.
A central objective in pharmacology is designing that do exactly what is intended and nothing else.
This section considers selectivity and specificity as quantitative pharmacological properties and illustrates how inadequate selectivity results in expectable adverse effects.
Therapeutic index is introduced as the formal expression of the margin between benefit and harm.
This also section considers clinical management in the context of narrow therapeutic index agents.1,2
![]() Selectivity
vs Specificity: Definitions and the Clinical Distinction
Selectivity
vs Specificity: Definitions and the Clinical Distinction
 |
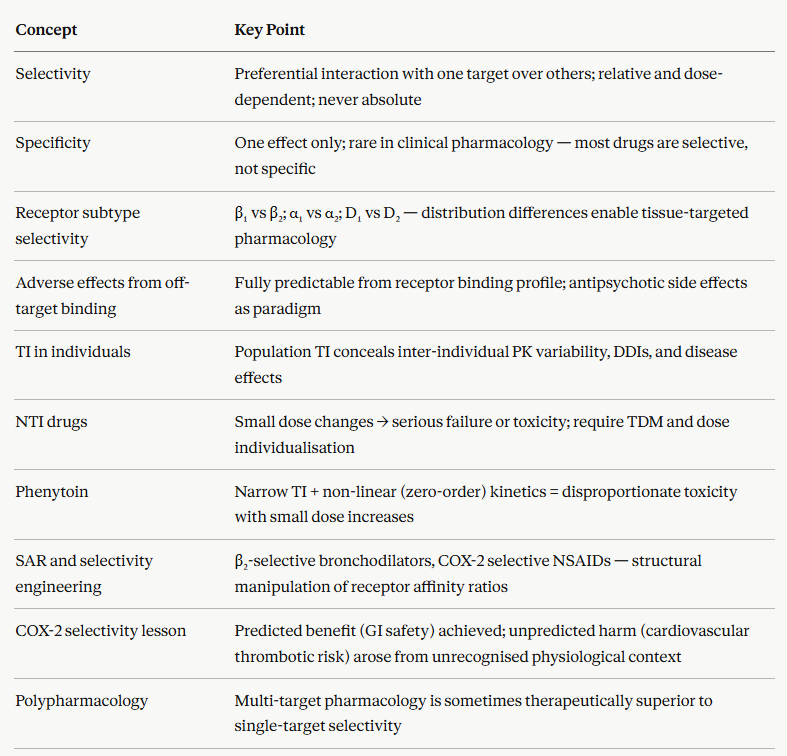
These two terms are sometimes used interchangeably in clinical conversation.
However, in pharmacology selectivity and specificity have distinct and precise meanings.3
Selectivity
Selectivity refers to the ability of a drug to interact preferentially with one receptor subtype, molecular target, or tissue over others.
Selectivity is always relative and dose-dependent.
A drug may be highly selective at therapeutic concentrations but at higher doses exhibits reduced selectivity as it interacts at lower-affinity sites.
Drugs should not be thought of as retaining very high selectivity independent of concentrations.4
Specificity
Specificity describes the ability of a drug to produce only a single, defined pharmacological effect since it interacts with only one type of receptor that initiates a singular type of downstream consequence.
True specificity is uncommon in clinical pharmacology.
Most drugs interact with multiple receptor subtypes or molecular targets, even if one predominates at therapeutic doses.3,4
The practical clinical implication of this distinction: prescribers should think in terms of selectivity windows rather than assuming any drug is specific.
The selectivity of a small molecule inhibitor is often significantly affected by the experimental dosage, because the dose-response curves for both the principal receptor target action and secondary target actions may be considerably different. NCBI
A drug that appears highly selective at one dose may produce significant off-target effects at a higher dose, a principle with direct relevance to toxicology and overdose management.
Receptor Subtype Selectivity: The Adrenergic System as a Model
The adrenergic receptor system is a good model for receptor subtype selectivity in clinical pharmacology, the adrenergic system illustrates both the therapeutic benefits of achieving selectivity and the clinical consequences if the appropriate selectivity is not met.
Adrenergic receptors are divided into two main classes, α and β each with multiple subtypes.
β-adrenoceptors are further divided into β1, predominantly cardiac: increases heart rate and contractility, and β2, predominantly airway and vascular smooth muscle mediating bronchodilation and vasodilation.
These subtypes share a common endogenous agonist, epinephrine and norepinephrine, but differ in their tissue distribution and G protein coupling.
Non-selective β-blockers (e.g. propranolol, nadolol) block both β1 and β2 receptors with equal affinity.
β1 blockade is therapeutically intended, reducing heart rate and blood pressure in hypertension, angina, and heart failure.
β2 blockade is typically not intended as such receptor blockade would result in bronchoconstriction in airway smooth muscle, clinically silent in patients with normal airways but potentially life-threatening in patients with asthma or COPD.
Non-selective β-blockers are absolutely contraindicated in asthma precisely because of this selectivity failure at β2.1,2
Cardioselective β-blockers (metoprolol, atenolol, bisoprolol)
These antagonists have a 10- to 100-fold higher affinity for β1 than β2 receptors.
At therapeutic doses of these agents, a meaningful β1 blockade with reduced β2 effects.
This selectivity is permits cautious use in patients with mild-to-moderate COPD who also need β-blockade for cardiac indications while recognizing that selectivity is never absolute and dose escalation progressively diminishes the β1/β2 selectivity ratio.4
α-adrenoceptor subtypes provide another instructive example.
α1-Adrenoceptors mediate vascular smooth muscle contraction.
α2-adrenoceptors mediate presynaptic inhibition of noradrenaline release (reducing sympathetic tone).
Drugs targeting the α1 subtype selectively (prazosin, doxazosin) are used in hypertension and benign prostatic hyperplasia
Drugs with mixed α1/α2 blockade (phentolamine) are used in pheochromocytoma crises, where blocking presynaptic α₂ auto-inhibition would worsen catecholamine release if not countered.
 |
Selectivity Across Receptor Families: Predicting Adverse Effects from Pharmacology
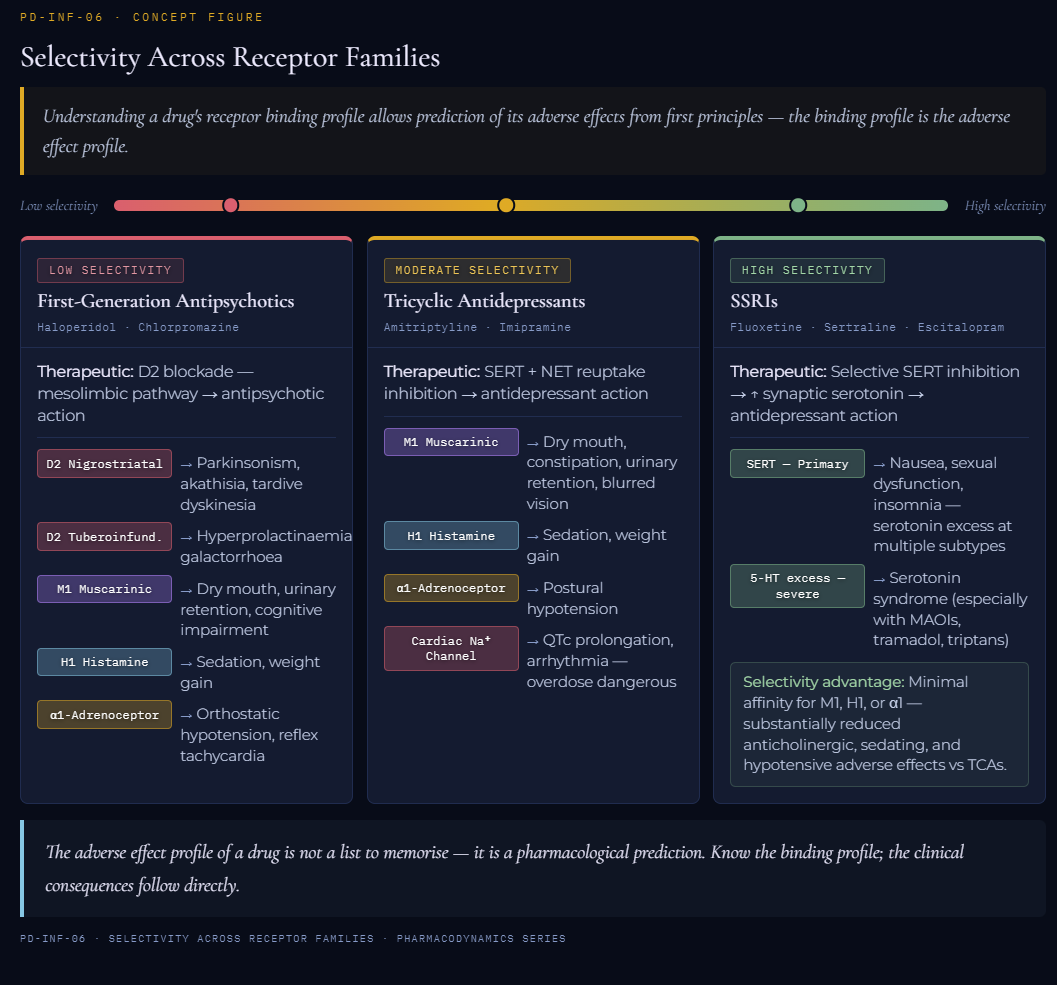 |
The principle that adverse drug effects follow predictably from off-target receptor interactions is important in clinical pharmacology concept.
Understanding a drug's receptor binding profile can anticipate its adverse effects from first principles.
Antipsychotics illustrate this relationship.
First-generation antipsychotics (haloperidol, chlorpromazine) produce their therapeutic effects through dopamine D2 receptor blockade in the mesolimbic pathway.
![]() Adverse
effects closely follow from recognition of additional receptor
interactions.
Adverse
effects closely follow from recognition of additional receptor
interactions.
D2 blockade in the nigrostriatal pathway → extrapyramidal side effects (parkinsonism, akathisia, tardive dyskinesia)
D2 blockade in the tuberoinfundibular pathway → hyperprolactinemia
Muscarinic M1 blockade → dry mouth, urinary retention, constipation, cognitive impairment
Histamine H1 blockade → sedation, weight gain
α1-Adrenoceptor blockade → orthostatic hypotension1,2
Tricyclic antidepressants (amitriptyline, imipramine)
These drugs produce antidepressant effects via serotonin and noradrenaline reuptake inhibition, but their binding to muscarinic, histaminergic, and α1-adrenergic receptors produces the anticholinergic side effect triad (dry mouth, constipation, urinary retention), sedation, and postural hypotension respectively.
Some tricyclic antidepressants interaction with cardiac sodium channels (quinidine-like effect) produces the QTc prolongation and elevated arrhythmia, making overdose particularly dangerous.
Serotonin Selective Reuptake Inhibitors (SSRIs) achieve greater selectivity by primarily targeting the serotonin transporter (SERT), with minimal affinity for muscarinic, histaminergic, or adrenergic receptors.
Accordingly, drugs belonging to this class of antidepressants exhibit substantially reduced anticholinergic and antihistaminergic adverse effects of tricyclics.
SSRI adverse effects (nausea, sexual dysfunction, insomnia, and in severe cases serotonin syndrome) are likely due to excessive serotonin at multiple receptor subtypes.
The Therapeutic Index Revisited: From Population Statistics to Individual Patients
 |
The therapeutic index (TI = TD50/ED50) may be viewed as a population-level statistic derived from quantal dose-response curves.
However, the TI concept applies with limitations at the level of the individual patient.
The fundamental limitation of the TI as a population statistic is that it conceals inter-individual variability.
Quantal dose-response curves for both efficacy and toxicity are population distributions since they reflect the spread of individual sensitivities around a median.
Two drugs may have identical TI ratios (identical TD50/ED50) but very different clinical safety profiles if their dose-response curves have different slopes or different degrees of overlap.9
Three factors compress the effective TI in individual patients
 |
Pharmacokinetic variability is the most important.
The same dose produces different plasma concentrations in different patients due to differences in body weight, renal function, hepatic CYP enzyme activity, protein binding, and genetic polymorphisms in drug-metabolising enzymes.
A patient who is a CYP2D6 poor metabolizer accumulates codeine's active metabolite morphine more slowly (or not at all), while a CYP2D6 ultrarapid metabolizer may generate toxic morphine concentrations from standard doses.
A patient with acute kidney injury receiving a renally cleared drug with a narrow TI is, pharmacokinetically, receiving a higher effective dose than prescribed.1,2
Drug-drug interactions at the pharmacokinetic level (CYP inhibition or induction) or pharmacodynamic level (additive or synergistic toxicity) effectively shift a patient's position on the dose-response curve without changing the prescribed dose.
Warfarin interacting with fluconazole (CYP2C9 inhibition) at a fixed warfarin dose produces warfarin plasma concentrations equivalent to a substantially higher dose, moving the patient toward the right tail of the toxicity distribution.8
Disease states alters both pharmacokinetics (renal clearance, hepatic metabolism, protein binding) and pharmacodynamics (receptor sensitivity, effector system responsiveness).
Elderly patients have reduced renal clearance, reduced hepatic CYP activity, and often increased receptor sensitivity.
Such factors effectively narrow the TI of drugs that are already narrow in young healthy adults.7
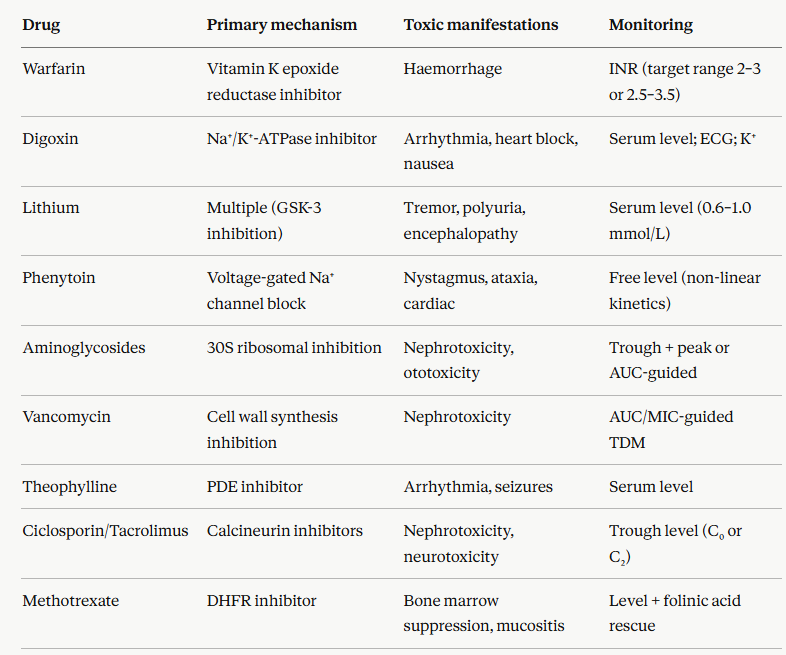
Narrow Therapeutic Index Drugs in Clinical Practice
Narrow therapeutic index drugs are defined by regulatory authorities as drugs where small differences in dose or blood concentration may lead to serious therapeutic failures and/or life-threatening adverse reactions.10
The FDA definition explicitly acknowledges that Narrow Therapeutic Index (NTI) drugs require tighter standards for bioequivalence and therapeutic drug monitoring than broad-TI drugs.10
![]() The clinical
characteristics of NTI drugs are consistent across the class:1,2,9
The clinical
characteristics of NTI drugs are consistent across the class:1,2,9
The dose-response curves for efficacy and toxicity overlap substantially.
As the consequence, there is no comfortable plateau of efficacy below the toxicity threshold.
Plasma concentration monitoring (therapeutic drug monitoring, TDM) is required to individualize dosing.
Dose adjustments must be made for changes in renal or hepatic function, body composition, interacting drugs, and age.
Small formulation differences (generic substitution, route changes, brand switches) can produce clinically meaningful concentration changes.
NTI drugs account for a disproportionate share of drug-related hospital admissions and adverse drug events.
 |
Structure-Activity Relationships and Selectivity
Selectivity is not an inherent property of a drug class.
Systematic modification of molecular structure permits identification of molecular structures which exhibit selectivity.
The relationship between molecular structure and pharmacological activity (the structure-activity relationship, SAR) is a vital element of rational drug design.1
Evolution of β2-selective bronchodilators illustrates this clearly.
Epinephrine (non-selective α and β agonist) was the first bronchodilator, but its α-mediated vasoconstriction and β₁-mediated tachycardia limited its clinical utility.
Structure-Activity studies showed that increasing the size of the substituent on the amine nitrogen of catecholamines progressively reduced α-receptor affinity while preserving or enhancing β affinity.
Additional structural modifications at the catechol ring produced β2 selectivity over β1.
Salbutamol (albuterol) and salmeterol are the clinical results of this program since these drugs exhibit sufficient β2/β1 selectivity ratios to achieve bronchodilation at doses that produce minimal cardiac stimulation.
Analogous SAR approaches produced:
β₁-selective blockers from the non-selective propranolol
H2-selective antihistamines from non-selective H1/H2 blockers, and
COX-2 selective NSAIDs (celecoxib, etoricoxib) from non-selective COX-1/COX-2 inhibitors.
In each case, the therapeutic rationale was the same: preservation of efficacy-associated interactions while limiting the toxicities.
The COX-2 selective NSAID story also illustrates the limits of this approach.
The rationale, that selective COX-2 inhibition would provide anti-inflammatory analgesia without the inhibition of gastroprotective COX-1.
An unanticipated consequence was that COX-2 selectivity in vascular endothelium shifted the prostanoid balance toward thromboxane A₂ (pro-thrombotic) and away from prostacyclin (anti-thrombotic) and this change led to increasing adverse cardiovascular event rates.
This is an example of an adverse effect, not predicted from the receptor pharmacology alone, because relevant physiological contexts were not fully appreciated at the time of development.1,2
Polypharmacology: When Selectivity Is Not the Goal
Maximum selectivity is not always the therapeutic ideal.
Intentional multi-target pharmacology may be beneficial.
Some of the most effective drugs in clinical use achieve their therapeutic effects precisely through interactions with multiple targets simultaneously, a property now termed polypharmacology.
Clozapine's superior efficacy in treatment-resistant schizophrenia, compared to more selective D2 antagonists, is thought to arise partly from its complex multi-receptor pharmacology (D2, D4, 5-HT2A, H1, α1, M1 among others).
Carvedilol's benefits in heart failure exceed what would be predicted from β-blockade alone, because it also has α1-blocking and antioxidant properties.
Amiodarone's antiarrhythmic efficacy arises from its actions on sodium, potassium, and calcium channels simultaneously, plus non-competitive β-adrenoceptor antagonism.5
 |
|
|
|
|
|
|
|
|