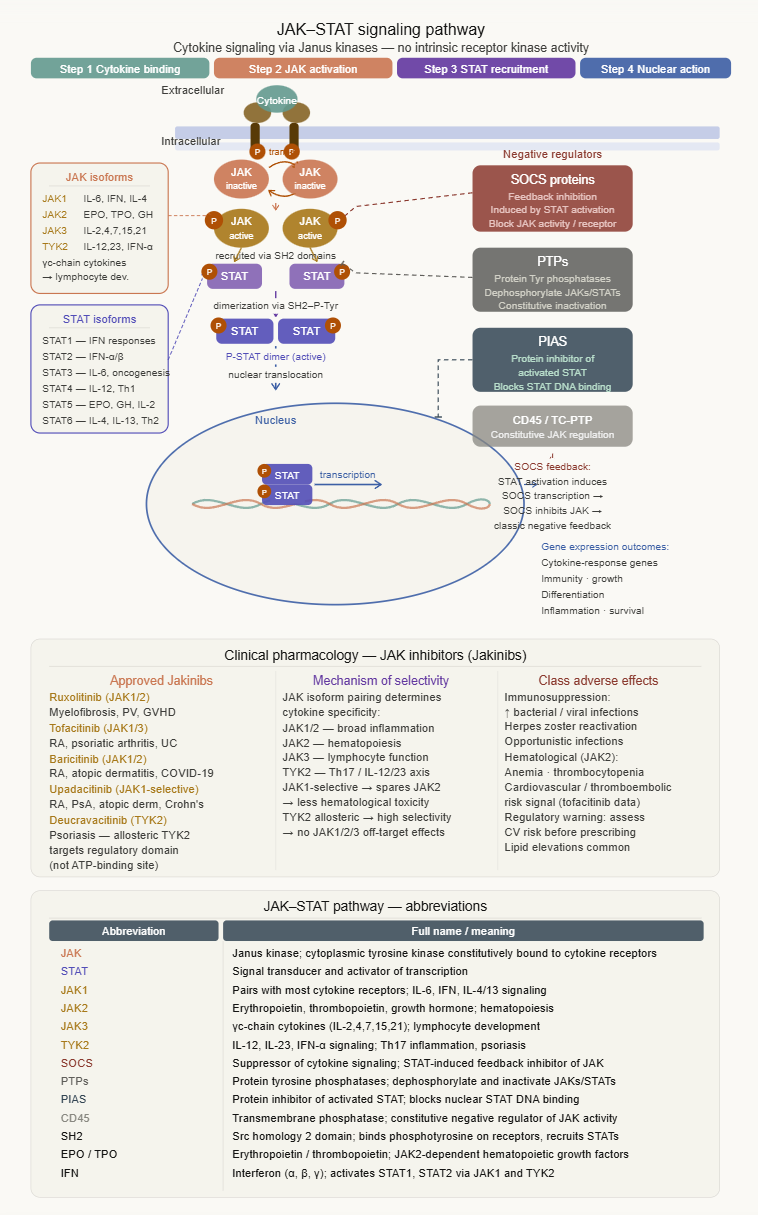
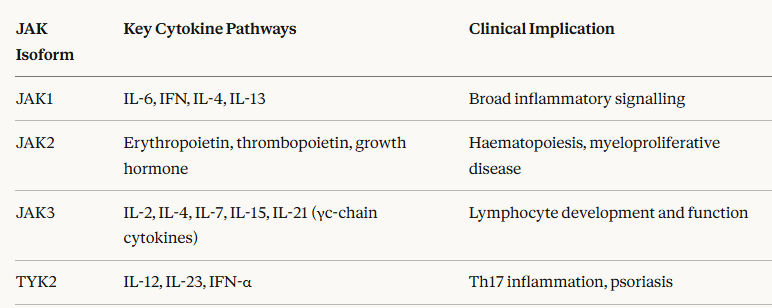
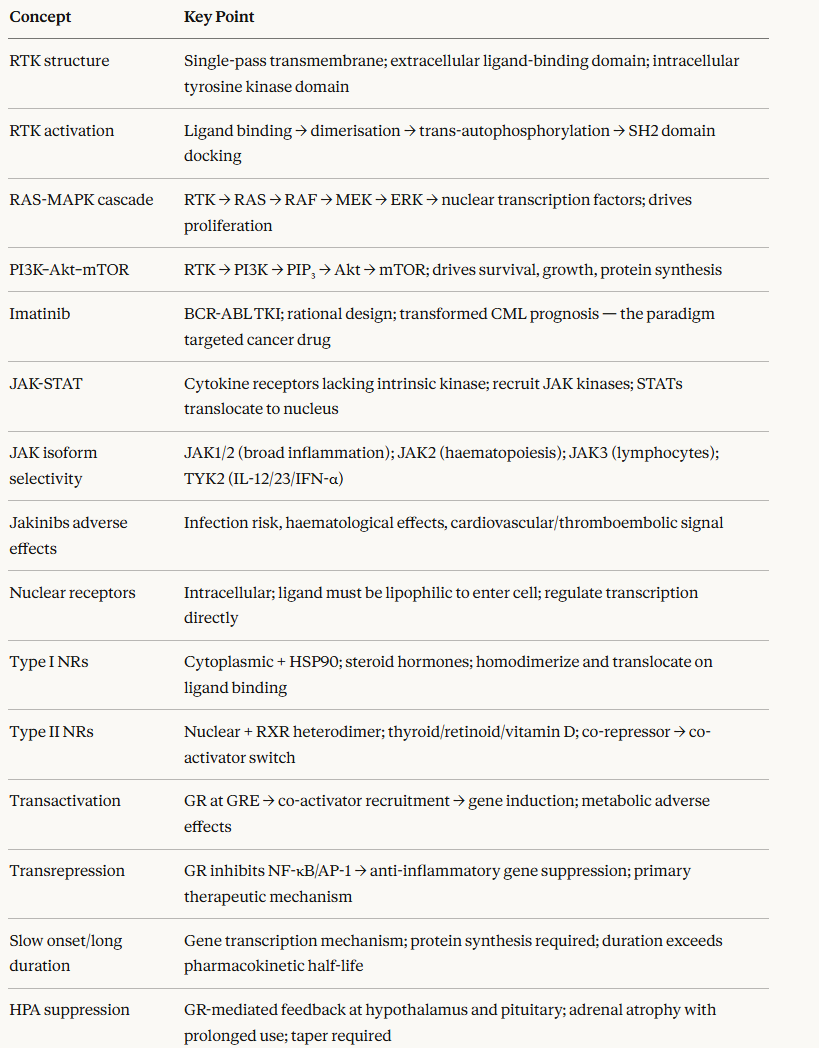
-
Brunton L Hilal-Dandan R Knollmann B, eds.
Goodman & Gilman's: The Pharmacological Basis of Therapeutics.
13th ed. McGraw-Hill; 2017.
-
Katzung B Vanderah T eds. Basic & Clinical
Pharmacology. 15th ed. McGraw-Hill; 2021.
-
Wintheiser G Silberstein P Physiology, Tyrosine
Kinase Receptors. In: StatPearls [Internet]. Treasure
Island, FL: StatPearls Publishing; 2023.
Click for Article
-
Lemmon M Schlessinger J Cell signaling by
receptor tyrosine kinases. Cell. 2010;141(7):1117–1134.
Click for Abstract
-
Maruyama IN. Mechanisms of activation of receptor
tyrosine kinases: monomers or dimers. Cells.
2014;3(2):304–330.
Click for Abstract
-
Babon J Lucet I Murphy J Nicola N Bhatt L The
molecular regulation of Janus kinase (JAK) activation.
Biochem J. 2014;462(1):1–13.
Click for Abstract
-
Rah B, Rather RA, Bhat GR, et al. JAK/STAT
signaling: molecular targets, therapeutic opportunities, and
limitations of targeted inhibitions in solid malignancies.
Front Pharmacol. 2022;13:821344.
Click for Abstract
-
Weikum ER, Liu X, Bhatt DL, Bhatt S, Bhatt D. The
nuclear receptor superfamily: a structural perspective.
Protein Sci. 2018;27(11):1876–1892. PMC6201731.
Click for Abstract
-
Aranda A, Pascual A. Nuclear hormone receptors
and gene expression. Physiol Rev. 2001;81(3):1269–1304.
Click for Abstract
-
Iqbal
N, Iqbal N. Imatinib: a breakthrough of targeted therapy in
cancer. Chemother Res Pract. 2014;2014:357027.
Click for Abstract