Medical Pharmacology Chapter 2 General Principles: Pharmacokinetics
|
|
|
|
|
|
|
-
-
-
-
Carrier-mediated transport is distinct from translocation of drugs across the membrane using channels.
-
Many drugs utilize specific membrane transporters for their passage across biological barriers.19
-
Facilitated diffusion involves carrier proteins that transport drugs down their concentration gradient without energy expenditure.
-
Active transport systems, including ATP-binding cassette (ABC) transporters and solute carrier (SLC) families, can move drugs against concentration gradients.21
-
These transporters exhibit substrate specificity, saturation kinetics, and potential for drug-drug interactions.
-
-
-
-
Facilitated diffusion is drug transport mediated by a carrier and the driving force is the electrochemical gradient associated with the drug.11,15,16
-
Facilitated Diffusion in Cell Membrane 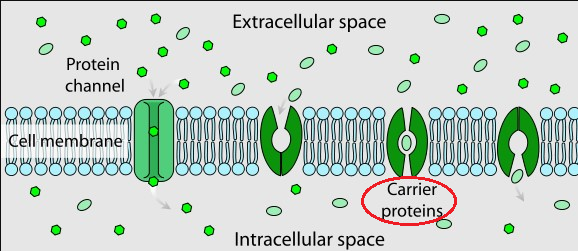
-
"Facilitated diffusion involves the use of a protein to facilitate the movement of molecules across the membrane.
-
In some cases, molecules pass through channels within the protein. In other cases, the protein changes shape, allowing molecules to pass through."
-
-
Attribution
-
LadyofHats, Public domain, via Wikimedia Commons
-
https://commons.wikimedia.org/wiki/File:Scheme_facilitated_diffusion_in_cell_membrane-en.svg
-
-
-
The electrochemical gradient consists of two elements:
-
(1) the difference in drug concentration across the membrane and the electrical gradient (difference in charge across the membrane).
-
(2) Specificity in such transport depends upon the carrier protein.
-
"Diagram of ion concentrations in charge across a semi-permeable cellular membrane" 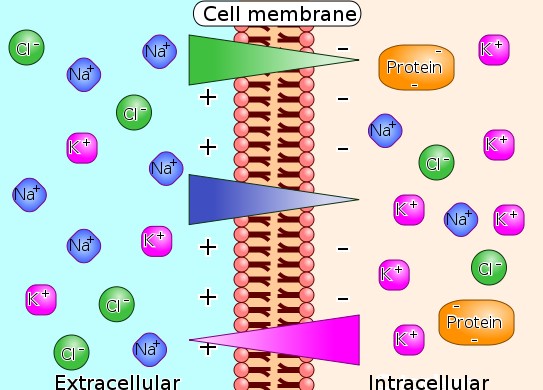
-
"Illustration of the way that differences in ion concentration on opposite sides of the membrane produces a voltage difference."
-
Attribution
-
Gibbs-donnan-en.svg: Biezlderivative work: Looie496,
-
CC BY-SA 3.0 <https://creativecommons.org/licenses/by-sa/3.0>, via Wikimedia Commons
-
-
-
-
-
The carrier protein is typically selective for a set of conformational structures.
-
The importance of the carrier is that absent such a carrier the translocation of the drug across the membrane would be much slower.
-
Drug binding to a carrier molecule induces a change in the complex confirmation which becomes energetically favorable for transporter across the membrane.
-
-
-
An example of a facilitated diffusion carrier is the organic cation transporter OCT1 coded by the gene SLC22A1.
-
This transporter, OCT1, not only facilitates transport of a physiological solute, thymine, but also is involved in transport of the drug metformin.
-
Metformin is useful in treating type II diabetes.11,15,16
-
-
OCT1 (gene: SLC22A1) is expressed in the liver and exhibits a relatively broad substrate specificity.17
-
OCT1 expression appears to correlate with drug responses.
-
Functionally defective OCT1 appears associated with drug resistance.17
-
-
-
 Members
of the OCT family (e.g. OCT1, OCT2, OCT3) are involved in renal
clearance of many drugs.18
Members
of the OCT family (e.g. OCT1, OCT2, OCT3) are involved in renal
clearance of many drugs.18 -
Transport of cations by OCTs is driven primarily by the membrane potential.
-
The OCTs noted above represent isoforms and these isoforms have overlapping substrates.
-
In addition, the tissue localization of these isoforms differ.
-
OCT1 is mainly associated in the sinusoidal or basolateral hepatocyte membranes.
-
Drugs and Endogenous Compounds and their Hepatocyte Transporters18 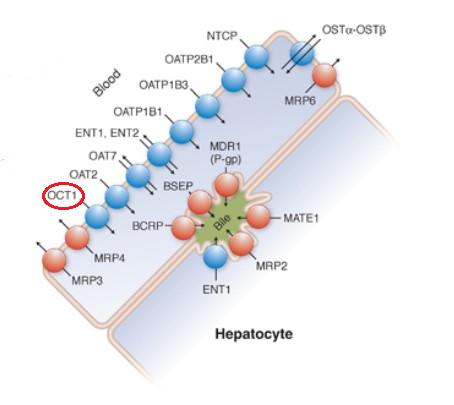
-
Attribution:
-
Adapted from Figure 5-1 from reference 18
-
Esroy BA Hoffmaster KA Chapter 5 Drug Transporters in Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy (Golan DE Armstrons EJ Armstong AW, eds) 4e Wolters Kluwer 2017.
-
-
-
-
OCT2 is mainly renal, localized in the kidney proximal tubule.
-
OCT3 is more broadly distributed in tissues including mainly, intestine, kidney, and liver.
-
OCT3 promotes intestinal absorption on one hand and liver and renal secretion of drugs on the other.
-
-
-
-
OCTs are
involved in uptake of many drugs including those belonging to
the sedative, antidepressant and β-antagonist categories.18
-
-
-
-
Endocytosis and Transcytosis22
-
Large molecules, including protein drugs and nanoparticle formulations, may cross membranes through endocytosis.
-
This process involves membrane invagination and vesicle formation, allowing internalization of drugs that cannot cross the lipid bilayer directly.
-
-
Transcytosis, where vesicles traverse the entire cell, is particularly important for drug delivery across the blood-brain barrier.22
-
-
-
-
The intestinal epithelium presents multiple pathways for drug absorption, including transcellular and paracellular routes.
-
Tight junctions between epithelial cells restrict paracellular transport to small, hydrophilic molecules.23
-
Drug efflux pumps like P-glycoprotein in the intestinal epithelium can limit absorption of certain substrates.24
-
-
-
-
The placental barrier separates maternal and fetal circulations, with implications for drug safety during pregnancy.
-
Most drugs cross the placenta by passive diffusion, though active transporters also play important roles.
-
The expression of drug transporters changes throughout pregnancy, affecting fetal drug exposure.26
-
-
Placental transfer refers to the passage of drugs from the maternal circulation across the placenta to the fetal circulation.
-
The placenta acts as a semi-permeable barrier, but most drugs can cross it to some extent.27
-
The rate and extent of drug transfer are influenced by several factors, including:
-
Lipid Solubility: Highly lipid-soluble drugs cross the placenta more readily than water-soluble drugs.28
-
Molecular Weight: Drugs with lower molecular weights (generally less than 500 Da) cross more easily. Larger molecules (e.g., insulin, heparin) typically have difficulty crossing.28
-
Ionization: Non-ionized (uncharged) drugs are more lipid-soluble and thus cross the placenta more readily than ionized drugs.28
-
Protein Binding: Only the unbound (free) fraction of a drug can cross the placenta. Highly protein-bound drugs will have limited transfer.28
-
Maternal and Fetal pH: The pH gradient between maternal and fetal blood can lead to "ion trapping," where a weakly basic drug becomes ionized in the more acidic fetal circulation, thus accumulating in the fetus.29
-
Transporters: The placenta expresses various transporters that may translocate drugs from the fetus back into the maternal circulation.
-
This activity may function as a fetal protective mechanism.30
-
-
-
-
-
-
-
-
-
Drug redistribution is an important pharmacokinetic process occurrings after initial drug distribution.
-
Drug redistribution refers to drug movement between different body compartments based on their physicochemical properties and tissue affinities.
-
This process significantly influences drug duration of action, accumulation patterns, and therapeutic outcomes.
-
Thiopental is an example of an induction anesthetic in which the drugs high lipid solubility combined with high blood flow to the brain result in a rapid increases in brain thiopental concentration soon after intravenous administration begins.35
-
However, the duration of action of thiopental anesthesia is relatively short because of rapid redistribution to other tissues ultimately adipose tissue which exhibits high capacity for lipophilic drugs.35
-
-
-
-
-
Mechanism of Drug Redistribution
-
Drug redistribution occurs through several interrelated mechanisms.
-
Following initial
distribution to highly perfused organs (brain, heart, liver, kidneys),
drugs subsequently redistribute to less perfused tissues such as muscle
and adipose tissue. 31
-
This redistribution is affected by:
-
Concentration gradients
-
Tissue binding affinities, and
-
Drug's lipophilicity.32,33
-
-
Principal driving forces for redistribution include:
-
Differences in blood flow between tissues
-
Varying tissue-to-plasma partition coefficients, and
-
Changes in protein binding over time.34
-
-
Highly lipophilic drugs tend to redistribute from the central nervous system to adipose tissue, while hydrophilic drugs may redistribute from highly perfused organs to muscle tissue.
-
Note that some parameters associated with distribution may be the same as parameters of importance in drug redistribution.
-
-
-
-
Updated
June 2025
|
|
|
|
|
|
|
|
|
References
|