|
|
|
|
|
|
|
Chapter 3: General Principles: Pharmacodynamics
|
|
|
|
|
|
|
|
|
|
|
Following the Discussion of the affinity in efficacy with respect to drug receptor interactions as well as dose response curves in the Hill equation, we now focus on what drugs do once bound to the receptor.
Maximum receptor activation
Partial receptor activation
Blocking receptor activation
Suppressing receptor activity to below baseline
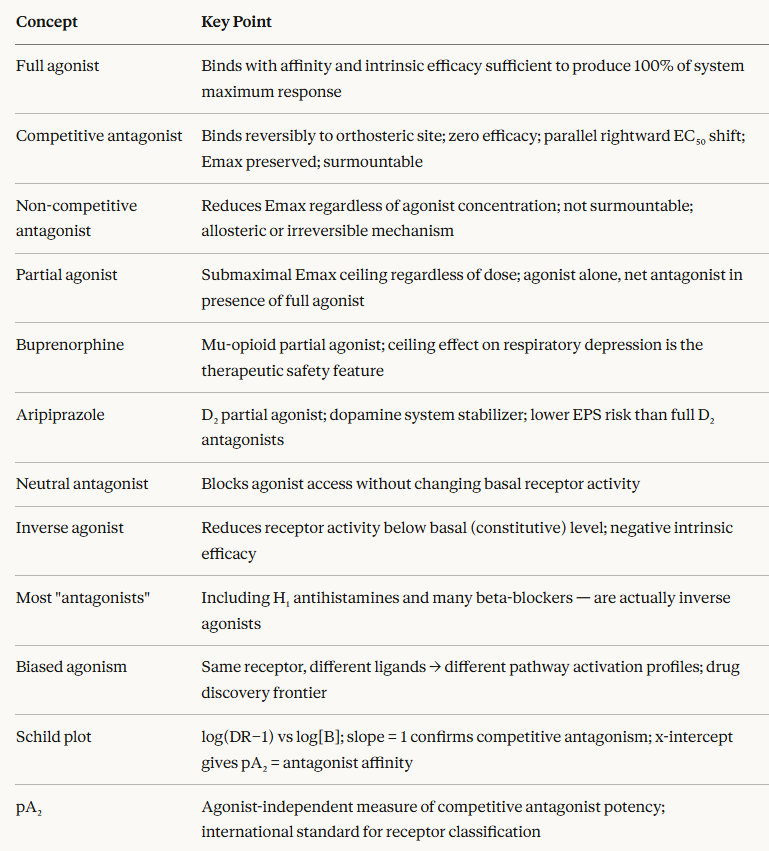
A full agonist is a ligand that, when it binds to a receptor, produces the maximum response of which the receptor-effector system is capable.
System is emphasized, since maximal efficacy is a property not of the drug in isolation but of the drug interacting with a specific receptor in a specific tissue.
A drug may be a full agonist in one tissue (producing 100% of maximum system response) and a partial agonist in another tissue with a less efficient signal transduction machinery, even though it occupies the same receptor in both.1,2
The molecular basis of full agonism is stabilization of fully active receptor conformation(s).
A full agonist has both high affinity for the receptor and a sufficiently high intrinsic efficacy that, when bound, it drives the receptor into the active state and promotes maximal downstream signalling through the effector system.
Clinical examples
Morphine at mu-opioid receptors acts as a full agonist for analgesia and respiratory depression; other agents acting as full agonists include:
Epinephrine at beta-adrenergic receptors
Isoproterenol at beta-adrenergic receptors
Acetylcholine at muscarinic and nicotinic receptors.
The dose-response curve of a full agonist reaches 100% of the system maximum.
Comparison of two full agonists for the same receptor may reveal differ substantially in potency (EC50) with one requiring a much lower concentration than the other to reach the same effect but not in maximal efficacy (Emax), which is identical for both since both can produce the system maximum.
A competitive antagonist is a ligand that binds reversibly to the same agonist binding site as an agonist, with affinity but zero intrinsic efficacy.
The competitive antagonist competes with the agonist for occupancy of the receptor as described by the principle of mass action.
Since binding is reversible and competitive, antagonist effects can always be overcome by increasing the agonist concentration sufficiently to displace the antagonist.
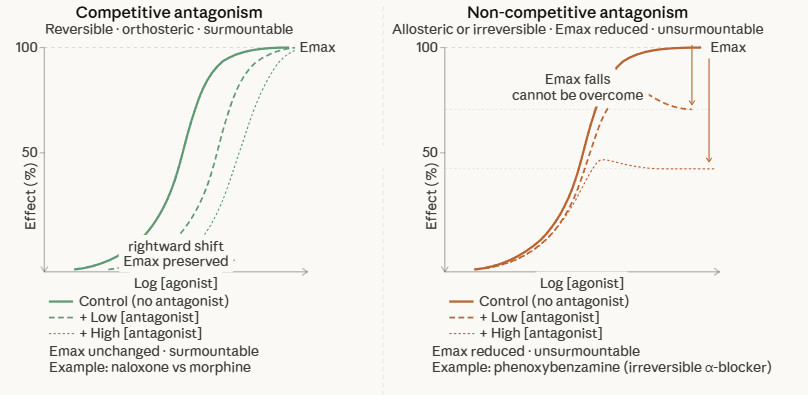
The defining pharmacological character of a competitive antagonist is the parallel rightward shift of the agonist's log dose-response curve in the presence of the antagonist, with no reduction in maximum efficacy (Emax):
Emax is preserved: maximum effect can still be achieved if sufficient agonist is present.
EC50 increases (potency is reduced) in proportion to the antagonist concentration.
The magnitude of the shift is quantified as the dose ratio (DR) which describes the fold increase in agonist concentration needed to restore the original EC50 in the presence of antagonist.
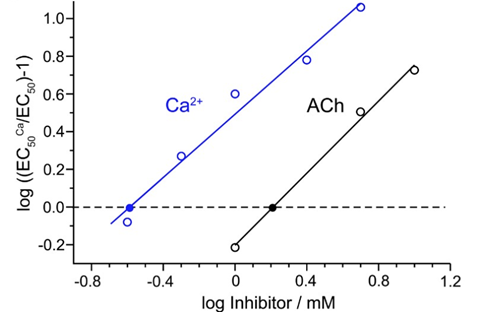
This EC50 shift is the quantitative basis of Schild analysis, developed by Hans Otto Schild in 1947 and 1949.3,4
Schild showed that for a competitive antagonist, the dose ratio follows a predictable mathematical relationship:
DR = 1 + [B]/KB
Where [B] is the concentration of antagonist and KB is its equilibrium dissociation constant (affinity for the receptor).
Rearranging: a plot of log (DR − 1) against the negative log of the antagonist concentration, i.e. the Schild plot, yields a straight line with a slope of unity for a simple competitive antagonist.
The x-intercept gives the pA2 value, which is the negative log of the antagonist concentration that doubles the agonist concentration required for the same response.5
 |
|
The Schild plot is clinically and experimentally important because:
(1) A slope of unity on the Schild plot confirms competitive antagonism; whereas, deviations indicate non-competitive mechanisms, receptor heterogeneity, or allosteric effects.
(2) The pA2 value provides a system-independent measure of antagonist affinity, likely identical regardless of which agonist is used, thus allowing comparison of antagonist potencies across different experimental conditions.
Clinical examples of competitive antagonists:
Naloxone at opioid receptors
Atropine at muscarinic receptors
Propranolol at beta-adrenergic receptors
Cetirizine and loratadine at H₁ histamine receptors;
Losartan at AT₁ angiotensin receptors.1,2
That competitive antagonism is surmountable by increasing agonist concentration has direct clinical significance.
In opioid overdose, naloxone competitively antagonizes mu-opioid receptors.
The duration of naloxone action (30–90 minutes) may be substantially shorter than the duration of the opioid, particularly for long-acting opioids like methadone or the lipophilic fentanyl.
Thus repeated naloxone administration or continuous infusion may be required to maintain effective antagonism while endogenous or exogenous opioids continue to compete for receptor occupancy.
A non-competitive antagonist reduces the maximum achievable response (Emax) regardless of agonist concentration.
This inhibition cannot be overcome by adding more agonist.
This inability to overcome distinguishes non-competitive from competitive antagonism and confers different clinical implications.
1. Irreversible antagonism
The antagonist binds covalently or with extremely slow dissociation to the same site as the agonist, permanently occupying receptors.
As the fraction of irreversibly blocked receptors increases, the maximum response progressively falls.
Phenoxybenzamine at α-adrenergic receptors is the archetypal example.
Phenoxybenzamine is used clinically in pheochromocytoma surgery because its "irreversibility" provides stable protection against catecholamine surges throughout the operative period, outlasting plasma drug levels.1,2
2. Allosteric non-competitive antagonism
Here the antagonist binds to a topographically distinct site (allosteric site), causing conformational changes in the receptor that reduce the efficacy of agonist-induced activation without preventing agonist binding.
Emax is reduced but the agonist can still bind (and EC50 may even be unchanged or reduced).
This mechanism underlies many modern drug interactions and is increasingly exploited in drug design.
On a log dose-response plot, the distinction is clear:
Competitive antagonist: parallel rightward shift, same Emax.
Non-competitive antagonist: rightward shift with Emax depression, or purely Emax depression with minimal EC50 change (for allosteric).
Surmountability is the key clinical test: can the effect of the antagonist be overcome by increasing agonist dose?
If yes, competitive.
If no, non-competitive.
Clinically, this means a non-competitive antagonist can reduce the maximum achievable effect even if agonist levels are very high, an important consideration for drugs with narrow therapeutic indices.
 |
A partial agonist binds to a receptor with affinity and produces receptor activation, but is incapable of producing the maximum response achievable by a full agonist, even at complete receptor saturation.
The Emax of a partial agonist is submaximal exhibiting a ceiling effect that persists regardless of dose.
Stephenson's (1956) concept of efficacy accounts for this observation.
The partial agonist has efficacy greater than zero in that it activates the receptor and produces a biological effect which is less than that of a full agonist since the stimulus per occupied receptor is not sufficient to activate the system maximum.
A partial agonist occupying all available receptors produces less stimulus than a full agonist occupying a fraction of them.
Partial agonist behavior depends upon context:
In the absence of a full agonist
Here the partial agonist behaves as an agonist, producing a submaximal yet real biological effect.
This property explains buprenorphine relief of withdrawal symptoms and cravings in opioid-dependent patients.
The partial agonist produces genuine mu-opioid receptor activation, providing analgesia and suppressing the motivational aspects of opioid withdrawal.
In the presence of a full agonist
In this instance the partial agonist behaves as a net antagonist.
By occupying receptors without producing the full stimulus of the endogenous agonist or a full agonist drug, it reduces the net response below what the full agonist alone would produce.
This characteristic explains why buprenorphine, given to a patient still receiving full opioid agonist doses, can precipitate acute withdrawal as the partial agonist displaces the full agonist from receptors while providing less activation.
This same mechanism is a basis of aripiprazole's antipsychotic efficacy:
As a partial D2 agonist, it reduces pathologically elevated dopamine signalling by competing with dopamine while providing reduced activation.
The ceiling effect of partial agonists is one of the most important safety properties in clinical pharmacology, and it is a pharmacodynamic property.
Buprenorphine (mu-opioid partial agonist):
The ceiling effect on respiratory depression is what makes buprenorphine intrinsically safer than full agonists like methadone or morphine in overdose.
Walsh et al. (1994) demonstrated in a dose-escalation study that buprenorphine doses up to 70 times the recommended analgesic dose were well tolerated by non-dependent subjects, with respiratory depression plateauing well below lethal levels.8
This safety profile is the pharmacological rationale for its use in opioid use disorder because its ceiling effect prevents the severe respiratory depression associated with full agonists overdose on one hand, while its high receptor affinity blocks the euphoric effect of subsequently administered full agonists on the other.
Aripiprazole (D2 receptor partial agonist)
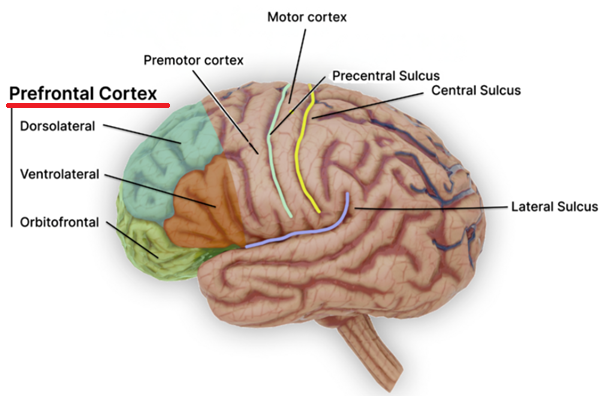
In brain regions with excessive dopaminergic activity, as in the mesolimbic pathway in schizophrenia, aripiprazole competes with dopamine and produces net receptor antagonism.
In brain regions exhibiting deficient dopaminergic activity, as in prefrontal cortex, implicated in schizophrenia negative symptoms, it provides partial agonist activation that partially corrects the deficit.
 |
|
This pharmacological profile with aripiprazole (Abilify) acting as a functional antagonist where dopamine is excessive and a functional agonist where it is deficient.
Accordingly, aripiprazole is classified as a dopamine system stabilizer.
Aripiprazole exhibits lower risk of drug-induced parkinsonism (extrapyramidal side effects including parkinsonism, dystonia, seizure, tardive dyskinesias) compared to full D2 antagonist antipsychotics.1,2
Varenicline (nicotinic acetylcholine receptor partial agonist)
Partial agonism at α4β2 nicotinic receptors provides sufficient activation to reduce nicotine withdrawal symptoms and cravings.
While reduced efficacy of varenicline, compared to nicotine and its competitive displacement of nicotine from the receptor prevent full reward reinforcement associated with cigarette smoking.
![]() The
partial agonist mechanism
represents a deliberately targeted therapeutic strategy with
the submaximal efficacy ceiling being the therapeutic feature.
The
partial agonist mechanism
represents a deliberately targeted therapeutic strategy with
the submaximal efficacy ceiling being the therapeutic feature.
The concept of the inverse agonist arose from an observation that contradicted the classical receptor model.
![]() Some drugs,
classified as antagonists based on their ability to block
agonist effects, actually
reduced
receptor activity below baseline in systems where the
receptor had constitutive (defined as spontaneous, ligand-independent)
activity.
Some drugs,
classified as antagonists based on their ability to block
agonist effects, actually
reduced
receptor activity below baseline in systems where the
receptor had constitutive (defined as spontaneous, ligand-independent)
activity.
Constitutive receptor activity is the ability of a receptor to adopt its active conformation and signal in the absence of any ligand Such behavior is a physiologically relevant phenomenon, particularly for G protein-coupled receptors (GPCRs) where populations of receptors exist in an equilibrium between inactive (R) and active (R*) conformations.
Some fraction of receptors may be in the R* state under basal conditions, generating a background level of signalling.7
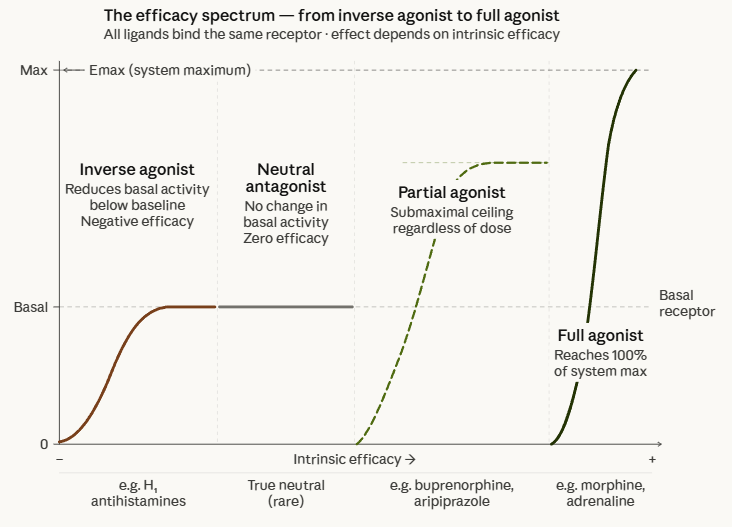
Within this two-state model of receptor activation, formalized by Black and Leff in 1983 and subsequently extended),6 ligands can be classified by their relative affinity for the active versus inactive receptor conformations:
Full agonist
High affinity for R* both stabilizes and expands the active receptor population, raising signalling above basal levels.
Neutral antagonist
Equal affinity for R and R* and occupies receptors without shifting the R/R* equilibrium; blocks agonist access without changing basal activity.
Inverse agonist
High affinity for R that stabilizes and expands the inactive receptor population, reducing basal signalling below baseline levels.
An inverse agonist produces the pharmacologically opposite effect of an agonist.
Where a full agonist at 100% efficacy (by convention) increases receptor activity, an inverse agonist produces negative efficacy which suppresses the receptor's constitutive activity.
Most drugs historically classified as "antagonists" are actually inverse agonists.
H1 antihistamines such as diphenhydramine, cetirizine, loratadine, fexofenadine are now classified as inverse agonists at the H1 receptor, reducing basal H1 receptor activity rather than simply blocking histamine access.7
β-blockers such as metoprolol and carvedilol show inverse agonism at β-adrenergic receptors in overexpressed systems.
The clinical consequences of this distinction at the receptor level may include the characteristic side effects of these drugs (sedation from H₁ inverse agonists) that would not be expected from a pure neutral antagonist, and may also explain some of the beneficial effects of β-blockers in heart failure beyond heart rate reduction.7
![]() The
distinction between neutral antagonism and inverse agonism
matters most in tissues with high constitutive receptor
activity, particularly disease states where receptor
mutations or receptor overexpression increase basal activity and
in drug withdrawal scenarios where removing an inverse agonist
allows constitutive activity to rebound.
The
distinction between neutral antagonism and inverse agonism
matters most in tissues with high constitutive receptor
activity, particularly disease states where receptor
mutations or receptor overexpression increase basal activity and
in drug withdrawal scenarios where removing an inverse agonist
allows constitutive activity to rebound.
 |
Biased Agonism: Functional Selectivity
Classical receptor pharmacology assumed that a drug produces a single pharmacological output determined by its receptor and its intrinsic efficacy.
|
G Protein-coupled receptors are the paradigm for biased agonism.
 |
|
Activated G-protein-coupled receptors signal through two primary pathways:
(1) G protein activation producing second messenger responses and
(2) β-arrestin recruitment involving receptor internalization, desensitization, and β-arrestin-mediated signalling.
![]() Different
agonists at the same receptor can preferentially
activate one pathway over the other by stabilizing
different active receptor conformations.9
Different
agonists at the same receptor can preferentially
activate one pathway over the other by stabilizing
different active receptor conformations.9
Kenakin and Christopoulos (2013) introduced a conceptual framework for quantifying this bias in a drug discovery setting, introducing the concept of biased signalling as both a measurable pharmacological quantity and a drug design target.9
The therapeutic promise of biased agonism has been most vigorously explored at opioid receptors.
Rationale
G protein signalling at mu-opioid receptors mediates analgesia
β-arrestin recruitment mediates receptor desensitization, tolerance, constipation, and may contribute to respiratory depression.
G protein-biased mu-opioid agonist might theoretically produce analgesia with reduced tolerance and respiratory depression risk.
Several G protein-biased opioid agonists have been developed such as oliceridine which received FDA approval in 2020.
Biased agonism represents an active and evolving area of drug discovery rather than a clinically established paradigm.9
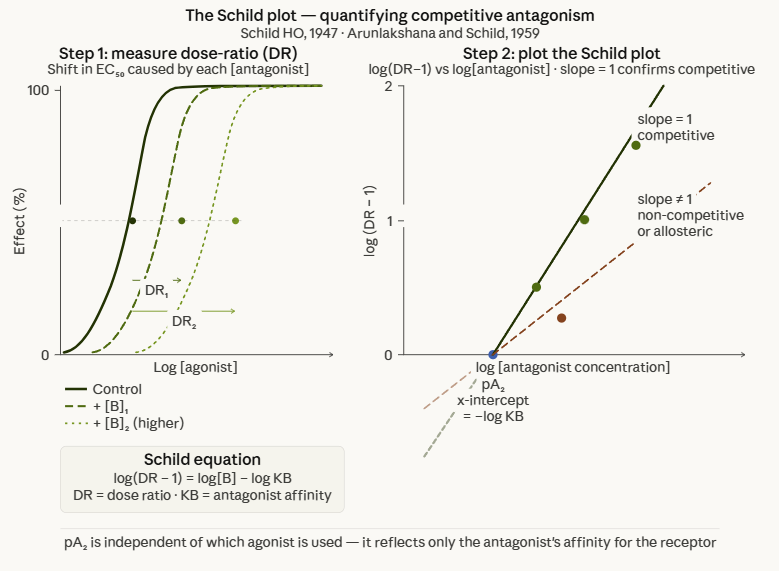
Schild Analysis: Quantifying Competitive Antagonism
The Schild plot is the gold-standard experimental method for
characterizing competitive antagonism quantitatively.
Construct an agonist concentration-response curve in the
absence of antagonist
Add a fixed concentration of antagonist and construct a
second agonist concentration-response curve
Repeat with multiple antagonist concentrations
For competitive antagonism: each addition of antagonist
produces a parallel rightward shift of the agonist curve,
preserving Emax.
The Schild equation (derived by Arunlakshana
and Schild, 1959)5
log(DR − 1) = log[B] − log KB
Where DR is the dose ratio (EC50 in presence of
antagonist / EC50 in absence), [B] is antagonist
concentration, and KB is the antagonist equilibrium dissociation
constant.
A plot of log(DR − 1) against log[B], the Schild plot,
yields a straight line with slope = 1 for simple competitive
antagonism.
The x-intercept (at log(DR − 1) = 0, i.e., DR = 2) gives the
pA₂ value which is the negative logarithm of
the antagonist concentration that doubles the required agonist
concentration.
The Schild plot shows log(DR−1)
on the y-axis against log[antagonist]
on the x-axis.
For a true competitive antagonist the
relationship is linear with slope = 1;
that is the solid green line, passing through the
x-axis intercept at the pA₂
point (= −log KB).
A slope significantly less than 1
(dashed red line) indicates non-competitive or
allosteric antagonism.
 |
|
|
|
|
|
|
|
|