|
|
|
|
|
|
|
Chapter 3: General Principles: Pharmacodynamics
|
|
|
|
|
|
|
Audio Overview: Pharmacodynamics: G-Protein-Coupled Receptors and Ion Channels I (Extended)
Audio Overview: Pharmacodynamics: G-Protein-Coupled Receptors and Ion Channels I (Extended) (Brief)
Signal Transduction I: G-protein-Coupled Receptors (GPCRs) and Ion Channels
| ; |
Introduction: Signal Transduction as the Mechanism of Pharmacodynamics
Pharmacodynamics, at its most fundamental level, is the study of signal transduction.
Signal transduction represents molecular mechanisms by which drug-receptor interactions are converted into cellular and physiological responses.
We have considered what happens at the receptor binding step and how affinity and efficacy determine the type and magnitude of receptor activation.
Now we take up how that receptor activation is translated into biological effects through the signaling machinery of the cell.
Signal transduction is not a passive relay.
Signal transduction is an amplifying, filtering, integrating, and regulatory system that specifies speed, magnitude, duration, and specificity of the cellular response to a drug.
Two receptor families, the G protein-coupled receptors (GPCRs) and ligand-gated ion channels (LGICs), represent the fastest and most clinically important signaling in pharmacology, operating on timescales from milliseconds to minutes.
Together they are the targets of the majority of clinically used drugs.1,2
Understanding signal transduction at a molecular level is helpful in, for example:
Explaining why beta-blockers are useful in heart failure despite reducing cardiac output in the short term
Explaining why benzodiazepines potentiate rather than mimic GABA
Explaining why ketamine's anaesthetic mechanism differs fundamentally from propofol's and
Explaining why tolerance to opioids and beta-agonists develops through receptor desensitization rather than receptor depletion.
|
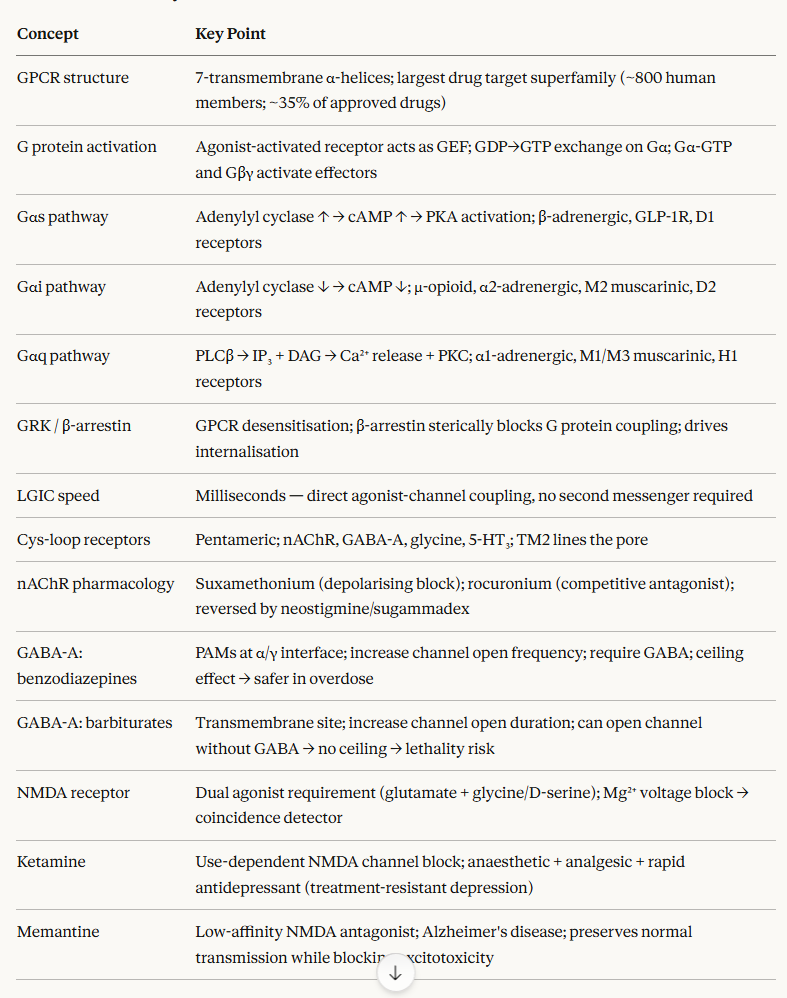
GPCRs: Structure and Mechanism of Activation
G protein-coupled receptors constitute the largest receptor superfamily in the human genome, with approximately 800 members.3
These systems respond to many signals including photons, odorants, taste molecules, ions, nucleotides, lipids, peptides, and proteins.
Abnormal function of G-protein-coupled receptor systems is implicated in many categories of human disease.
About 35% of all approved drugs act through GPCRs, making them the single most therapeutically important class of drug target.4
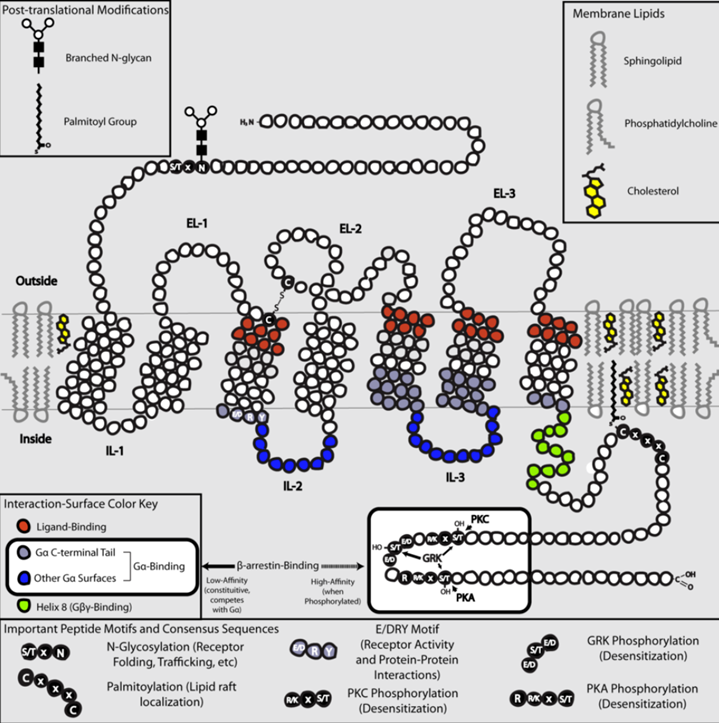
![]() All GPCRs
share a conserved structural core consisting of seven
hydrophobic transmembrane α-helices (TM1–TM7) connected by
three extracellular loops (ECL1–3) and three intracellular loops
(ICL1–3), with an extracellular N-terminus and an intracellular
C-terminus.3
All GPCRs
share a conserved structural core consisting of seven
hydrophobic transmembrane α-helices (TM1–TM7) connected by
three extracellular loops (ECL1–3) and three intracellular loops
(ICL1–3), with an extracellular N-terminus and an intracellular
C-terminus.3
This arrangement creates a barrel-like structure within the lipid bilayer, with the orthosteric (main binding site, distingushed from allosteric binding sites) ligand-binding domain typically embedded within the transmembrane bundle (for small molecules) or formed by the extracellular loops and N-terminus (for peptides and proteins).
 |
|
|
|
|
![]() Agonist
binding to the orthosteric site (natural, non-allosteric
site) induces conformational changes in the
transmembrane bundle, particularly an outward movement of
TM5 and TM6 sites on the intracellular face.
Agonist
binding to the orthosteric site (natural, non-allosteric
site) induces conformational changes in the
transmembrane bundle, particularly an outward movement of
TM5 and TM6 sites on the intracellular face.
The conformational change opens an intracellular cavity capable of accommodating the α-subunit of a heterotrimeric G protein.
This structural rearrangement is the key event that couples extracellular ligand binding to intracellular signal generation.3,4
Heterotrimeric G proteins consist of three subunits: Gα, Gβ, and Gγ. In the inactive state, Gα is bound to GDP and associated with the Gβγ dimer.
Agonist-activated GPCR acts as a guanine nucleotide exchange factor (GEF), catalysing the exchange of GDP for GTP on the Gα subunit. GTP-loaded Gα dissociates from Gβγ, and both the Gα-GTP and the free Gβγ dimer engage downstream effectors.3
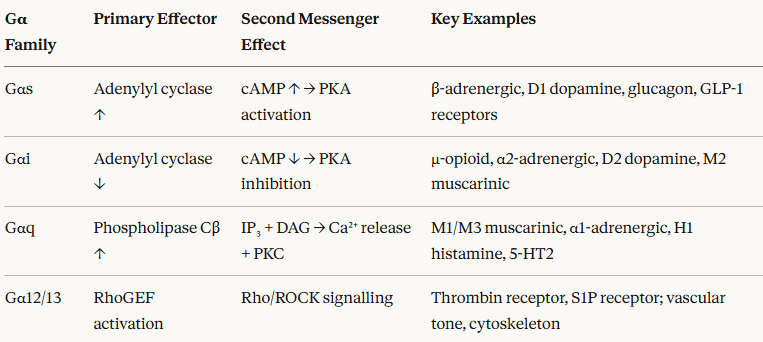
The four major Gα families and their primary second messenger effects
 |
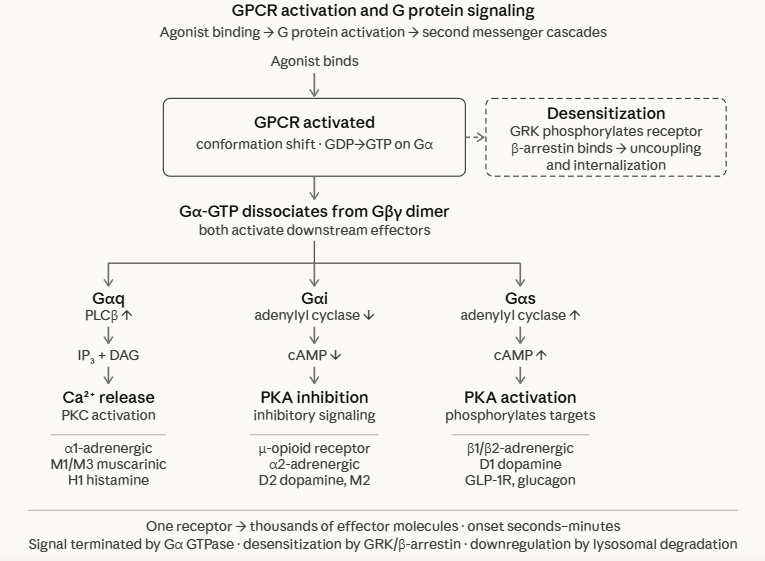 |
![]() The
duration of G protein signalling is terminated by the intrinsic
GTPase activity of Gα, which hydrolyses GTP back to GDP,
allowing reassociation with Gβγ and return to the inactive
state.
The
duration of G protein signalling is terminated by the intrinsic
GTPase activity of Gα, which hydrolyses GTP back to GDP,
allowing reassociation with Gβγ and return to the inactive
state.
RGS proteins (Regulators of G protein Signalling) dramatically accelerate this GTPase activity and are an important physiological brake on GPCR signalling.
cAMP–PKA Pathway (Gs Signalling)
The adenylyl cyclase–cAMP–protein kinase A (PKA) cascade is the most extensively characterised GPCR signalling pathway and the mechanism of action for beta-adrenergic agonists, glucagon, GLP-1 receptor agonists, and many other clinically crucial drugs.
Agonist activates Gs-coupled receptor (e.g. β₁-adrenoceptor, GLP-1R)
Gαs-GTP activates adenylyl cyclase, an integral membrane enzyme that converts ATP to cyclic AMP (cAMP)
cAMP activates protein kinase A (PKA) by binding to its regulatory subunits, releasing active catalytic subunits
PKA phosphorylates multiple substrate proteins, producing diverse cellular effects depending on cell type.
Clinical application: Cardiac pharmacology
In cardiac myocytes, β₁-adrenoceptor activation (Gs → cAMP → PKA) phosphorylates L-type calcium channels, ryanodine receptors, phospholamban, and myosin-binding protein C.
This activation results in positive inotropy (increased force), positive chronotropy (increased rate), and positive lusitropy (enhanced relaxation).
Beta-blockers, competitive antagonists at β1-adrenoceptors, attenuate all of these effects, which may be helpful in heart failure, angina, and arrhythmia management.
The paradox of beta-blocker use in heart failure, where reducing cardiac output might seem counterproductive, is understood by the pathological consequences of:
Sustained Gs/cAMP signaling in the failing heart leading to receptor desensitisation
Down-regulation
Myocyte apoptosis, and
Adverse cardiac remodelling.
Beta-blockade, by limiting this chronically elevated cAMP signal, allows receptor upregulation and partial restoration of β1-adrenoceptor sensitivity resulting in long-term benefit that outweighs short-term hemodynamic compromise.1,2
 |
|
|
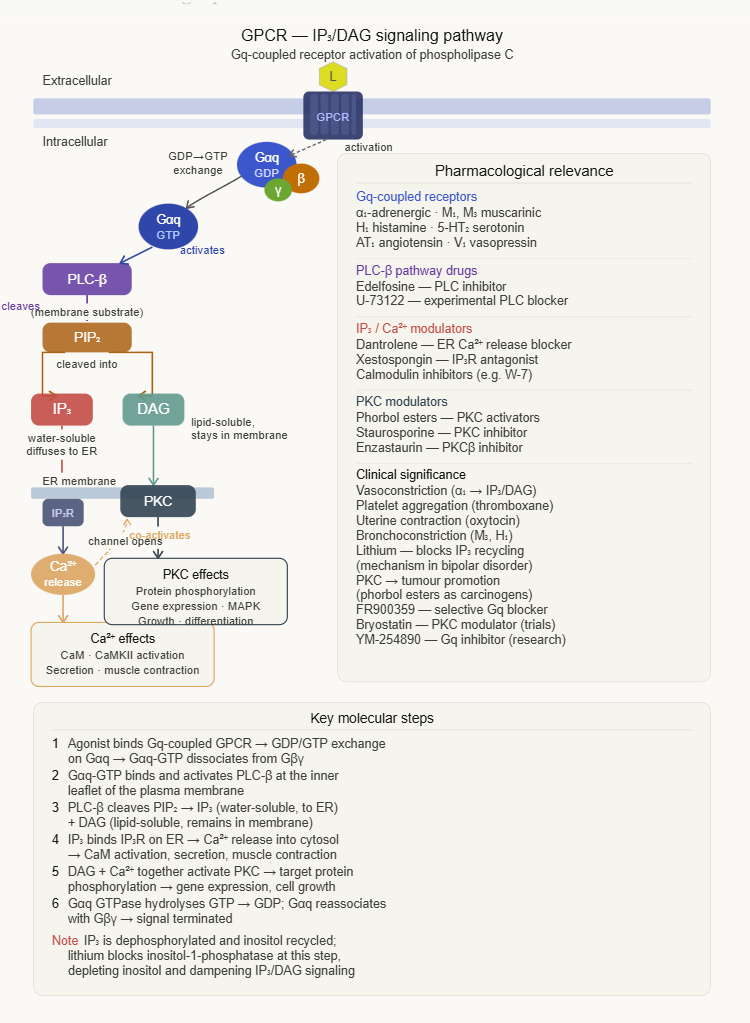
Gq-coupled receptor activation is associated with a different second messenger cascade, with calcium as the central signaling molecule.
(1) Agonist activates Gq-coupled receptor (e.g. α1-adrenoceptor, M1/M3 muscarinic, H1 histamine)
(2) Gαq-GTP activates phospholipase Cβ (PLCβ)
(3) PLCβ (phospholipase-Cβ) cleaves phosphatidylinositol 4,5-bisphosphate (PIP₂) into two second messengers:
Inositol trisphosphate (IP₃)
Diffuses to the endoplasmic reticulum, binds IP₃ receptors, triggers Ca2+ release into cytoplasm
Diacylglycerol (DAG)
Remains membrane-bound, activates protein kinase C (PKC)
(4) Elevated cytoplasmic Ca2+ activates calmodulin, which in turn activates multiple kinases (such as CaM kinase II, myosin light chain kinase) and other effectors.
(5) PKC phosphorylates a range of target proteins, modulating transcription, contractility, and secretion.
α1-adrenoceptor agonists (phenylephrine, noradrenaline) → Gq → IP3/Ca2+ → smooth muscle contraction → vasoconstriction
M1/M3 muscarinic agonists: → Gq → IP3/Ca2+ → glandular secretion, smooth muscle contraction (gut motility, bronchospasm); M3 antagonism (e.g. tiotropium) prevents this in COPD
H1 antihistamines block Gq-coupled H3 receptors, preventing the Ca2+ release that mediates histamine-induced itch, bronchoconstriction, and mucus secretion.1,2
GPCR Desensitisation, Internalization, and the β-Arrestin Pathway
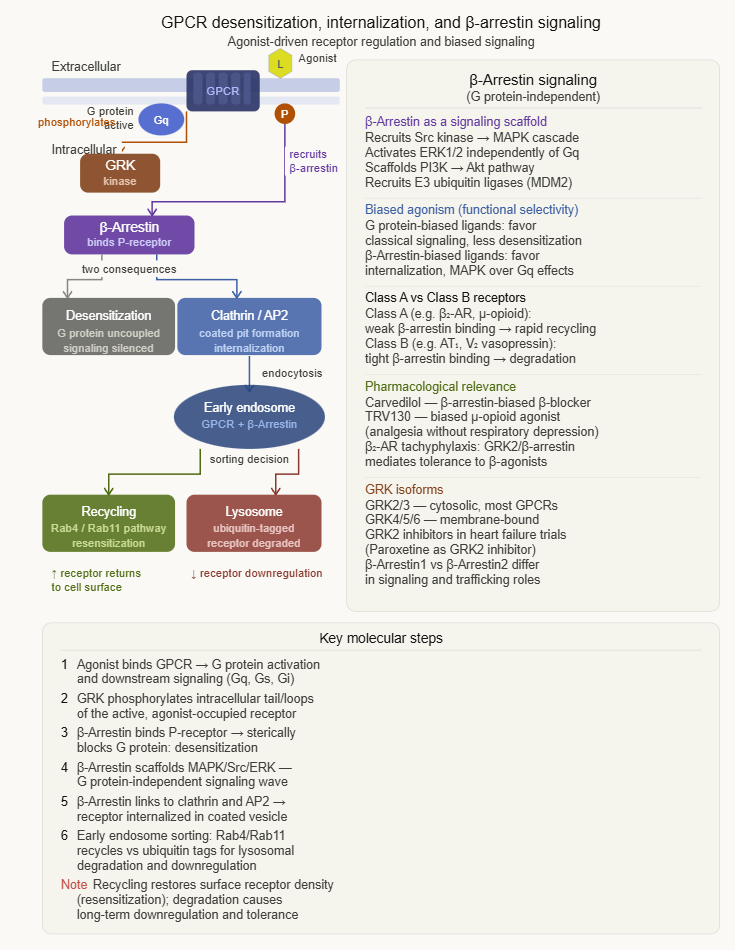 |
Sustained agonist stimulation of GPCRs triggers a multi-layered regulatory response that progressively attenuates receptor signaling.
This process is central to understanding tolerance, tachyphylaxis, and paradoxical effects of receptor regulation in disease states.
Three overlapping mechanisms of desensitisation
1. Receptor phosphorylation (seconds)
GPCR kinases (GRKs) phosphorylate serine and threonine residues on the intracellular loops and C-terminus of the agonist-occupied receptor.
This phosphorylation does not directly impair G protein coupling but creates docking sites for β-arrestin proteins.
2. β-Arrestin binding (seconds to minutes)
β-Arrestins (β-arr1 and β-arr2) bind to GRK-phosphorylated receptors, physically sterically blocking G protein access to the receptor.
This process is called uncoupling or homologous desensitization and represents the primary mechanism of rapid desensitization as well as the pharmacological basis of tachyphylaxis to β-agonist bronchodilators and other short-acting agonists.
3. Receptor internalzation (minutes to hours)
β-Arrestin-bound receptors are directed into clathrin-coated pits and internalized by endocytosis.
Internalized receptors are either recycled back to the cell surface thus restoring sensitivity or directed to lysosomes for degradation. Degradation reduces total receptor number resulting in downregulation.
Prolonged agonist exposure drives downregulation, requiring new receptor synthesis for full recovery, representing the molecular basis of opioid tolerance.3,4
β-Arrestin as a signaling molecule
Beyond desensitization, β-arrestin itself serves as a scaffold for signaling complexes independent of G proteins, activating kinase cascades including ERK1/2 and src.
This characteristic is the structural basis for biased agonism described earlier in which agonists that preferentially engage β-arrestin signaling over G protein signaling produce pharmacologically distinct profiles.
Ligand-Gated Ion Channels: Structure and Signaling Principles
Ligand-gated ion channels (LGICs) are the second major class of signal transducing receptors responsible for fast neurotransmission.
Ligand-gated ion channels distinguishing feature, speed, is a direct architectural consequence in that agonist binding directly opens an ion channel within the same protein, without requiring second messenger intermediaries.6,9
Structural organization
LGICs are oligomeric transmembrane proteins in which the ligand-binding domain and the ion channel are both components of the same macromolecular assembly.
The two major LGIC superfamilies differ in their quaternary organization
Cys-loop receptors (pentameric LGICs)
Cys-loop receptors are named for a disulphide-bond-formed loop in the extracellular domain.
These receptors are pentamers with five subunits arranged symmetrically around a central ion-conducting pore.
Each subunit contains a large extracellular N-terminal domain (where agonist binds at the subunit interfaces) and four transmembrane helices (TM1–TM4), with TM2 from each subunit lining the central pore.
The Cys-loop family includes:
Nicotinic acetylcholine receptors (nAChR) — cation-selective (Na+ K+, Ca2+) → excitatory
GABA-A receptors: anion-selective (Cl⁻) → inhibitory
Glycine receptors: anion-selective (Cl⁻) → inhibitory
5-HT3 receptors: cation-selective → excitatory
Ionotropic glutamate receptors (tetrameric LGICs)
Tetramers with a structurally distinct transmembrane domain topology including a re-entrant pore loop (analogous to potassium channel structure) that lines the selectivity filter.
Three major classes:
AMPA receptors (fast excitatory transmission)
NMDA receptors (coincidence detectors, Ca²⁺-permeable), and
Kainate receptors
Gating mechanism
Agonist binding to the extracellular domain triggers a conformational change, involving rotation and/or rearrangement of the transmembrane helices, that opens the channel pore.
This conformational coupling between the binding site and the channel gate occurs within microseconds, accounting for the extraordinary speed of LGIC-mediated signaling (milliseconds to tens of milliseconds to peak response).9
Nicotinic Acetylcholine Receptors: The Prototype LGIC
The muscle-type nicotinic acetylcholine receptor (nAChR) is the best-characterized LGIC and the prototype for the Cys-loop superfamily.
Nicotinic acetylcholine receptor pharmacology is clinically important at the neuromuscular junction (NMJ), mediating motoneuron signal to skeletal muscle.
Structure
Muscle nAChR (Nicotinic acetylcholine receptor) is a heteropentamer of subunit composition α₂βγδ (fetal) or α₂βεδ (adult), with the two acetylcholine binding sites located at the interfaces of the α-subunit with the adjacent γ (or ε) and δ subunits.
Both ACh binding sites must be occupied simultaneously to open the channel efficiently representing a requirement for dual occupancy underlying cooperative gating kinetics (Hill coefficient > 1) of the receptor.
Pharmacological modulation
Depolarizing neuromuscular blockers (suxamethonium/succinylcholine)
These agents are nAChR agonists, causing initial fasciculation followed by prolonged depolarization blockin which the channel initially opens, then persistent depolarization causes desensitization and failure of action potential generation.
Ultra-short duration due to rapid hydrolysis by plasma cholinesterase.
Drug belonging to this category are important in anesthesia,used for rapid sequence induction.
Non-depolarizing neuromuscular blockers such as rocuronium, vecuronium, atracurium act as competitive antagonists at nAChR.
These agents bind at ACh bind sites but without activating the channel thus preventing ACh-mediated depolarization.
Blocking action of these non-depolarizing antagonists may be reversed by acetylcholinesterase inhibitors such as neostigmine.
Sugammadex reverses rocuronium/vecuronium by direct encapsulation.
Organophosphates and carbamates
Some agents belonging to these categories inhibit acetylcholinesterase, causing ACh accumulation → prolonged nAChR stimulation → depolarization block which when combined with muscarinic overstimulation lead to Salivation, Lacrimation, Urination, Defaecation, GI distress, Emesis or SLUDGE).
Treatment is atropine which, as a muscarinic antagonist blunts the effects of excess ACh along with pralidoxime that may reactivate phosphorylated acetylcholinesterase.1,2
GABA-A Receptors and CNS Depressants
The GABA-A receptor is the primary mediator of fast inhibitory neurotransmission in the CNS and the target of some of the most widely prescribed drug classes in clinical medicine.
Structure
GABA-A receptors are heteropentamers, with the most prevalent brain form comprising 2α + 2β + 1γ subunits (most commonly α1β2γ2).
GABA orthosteric (non-allosteric) binding sites are located at the β+/α− subunit interfaces (two sites per pentamer).
The channel is anion-selective (Cl⁻), and GABA-mediated channel opening causes Cl⁻ influx, membrane hyperpolarization, and inhibition of action potential generation.7
Multiple allosteric binding sites and their pharmacology
Benzodiazepine site (α+/γ− interface)
Benzodiazepines (diazepam, lorazepam, midazolam) bind as positive allosteric modulators (PAMs) at the extracellular interface between the α and γ subunits.
These drugs do not directly open the Cl⁻ channel as they require GABA to be present.
Their modulatory effect is to increase the frequency of channel opening in response to GABA.
This is by an allosteric mechanism, not direct agonism and help to explain why benzodiazepines have a ceiling effect on CNS depression and are intrinsically safer in overdose than barbiturates.7
Barbiturate site (transmembrane domain)
Barbiturates (phenobarbitone, thiopentone) bind at transmembrane domain sites distinct from the benzodiazepine site.
At low concentrations the barbiturates increase the duration of channel opening, complementary to benzodiazepines' frequency effect.
At high concentrations they can directly open the Cl⁻ channel independently of GABA which explains barbiturates' lack of a ceiling effect and their greater lethality in overdose compared to benzodiazepines.
![]() This
is also why barbiturate–benzodiazepine combinations produce
supra-additive CNS depression.7
This
is also why barbiturate–benzodiazepine combinations produce
supra-additive CNS depression.7
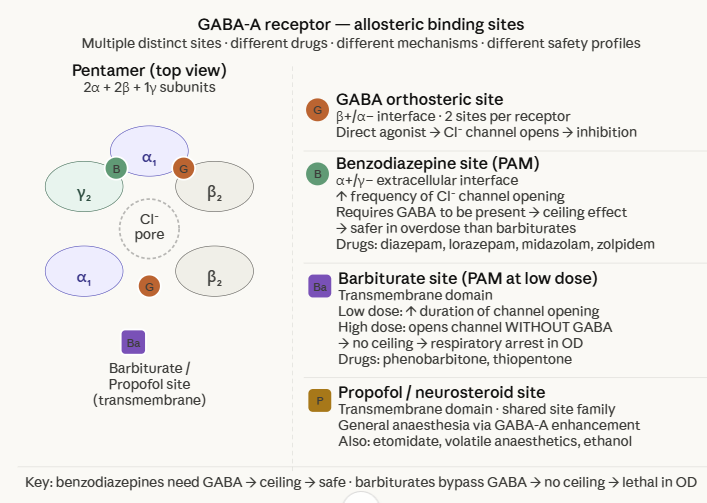 |
Neurosteroid site (transmembrane domain)
Endogenous neurosteroids (allopregnanolone, THDOC) and exogenous agents including brexanolone, the first neurosteroid drug approved for postpartum depression act at transmembrane sites, enhancing GABA-A function.
Fluctuation of neurosteroid levels across the menstrual cycle and postpartum period underlies some mood disorders whose pathophysiology centers on altered GABA-A sensitivity.
Propofol and volatile anesthetics (transmembrane domain)
Propofol, etomidate, volatile anesthetics (sevoflurane, isoflurane), and ethanol all enhance GABA-A function through transmembrane domain interactions.
![]() This
convergence of chemically diverse general anesthetics on the
GABA-A receptor provides strong support for GABA-A modulation as
a primary mechanism of general anesthesia.7
This
convergence of chemically diverse general anesthetics on the
GABA-A receptor provides strong support for GABA-A modulation as
a primary mechanism of general anesthesia.7
NMDA Receptors: Coincidence Detection and Clinical Pharmacology
The NMDA (N-methyl-D-aspartate) receptor is an ionotropic glutamate receptor with a unique voltage-dependent gating mechanism that makes it a molecular coincidence detector.
This characteristic underlies both its physiological role in learning and memory and the mechanism of action of several clinically important drugs.
Unique activation requirements
NMDA receptors require simultaneous binding of two different agonists: glutamate (at the GluN2 subunit) and a co-agonist, either glycine or D-serine (at the GluN1 subunit).
Both sites must be occupied for the channel to open.
Uniquely among LGICs, the NMDA channel is subject to voltage-dependent Mg²⁺ block: at resting membrane potential (around −70 mV), Mg²⁺ ions occlude the channel pore even when both agonist sites are occupied.
Only when the membrane is depolarized (e.g. during an action potential or nearby synaptic activity) is the Mg²⁺ block relieved.
This observation suggests that NMDA receptors open only when the cell is already active.
These receptors apparently detect coincident presynaptic glutamate release AND postsynaptic depolarisation.6
This coincidence detection is the cellular mechanism of long-term potentiation (LTP), the synaptic model of learning and memory. The Ca2+ influx through activated NMDA receptors triggers the kinase cascades that strengthen the synapse.
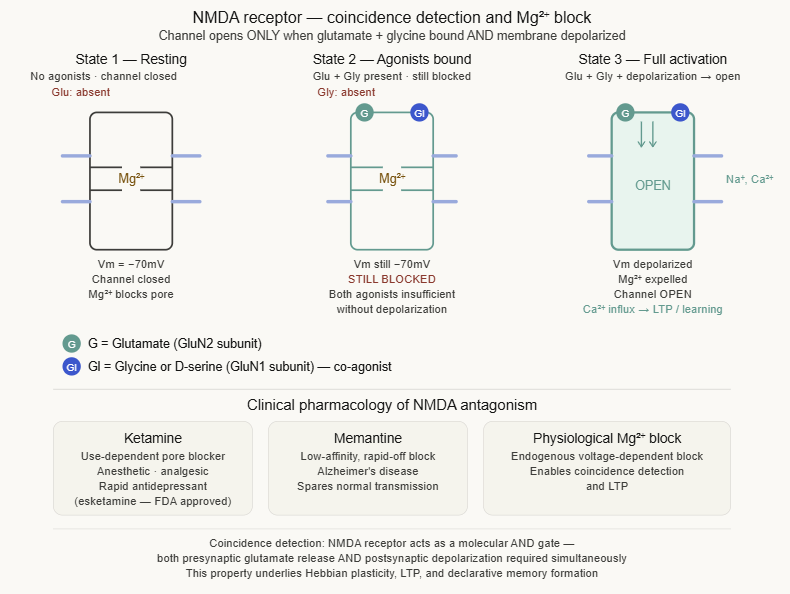 |
Clinical pharmacology of NMDA receptor antagonism
Ketamine (non-competitive NMDA antagonist): binds within the channel pore in a use-dependent manner in that the channel must open for ketamine to enter.
Anesthetic, analgesic, and dissociative properties all arise from NMDA blockade.
Sub-anesthetic doses produce analgesia and recently demonstrated rapid antidepressant effects noting that IV ketamine and intranasal esketamine are now approved for treatment-resistant depression.
Antidepressant mechanism likely involves downstream synaptogenesis secondary to NMDA blockade.
Memantine (NMDA antagonist)
Low-to-moderate affinity, rapid-off kinetics and the channel block is surmountable under normal physiological activity.
Approved for moderate-to-severe Alzheimer's disease where it may block pathological NMDA overactivation (excitotoxicity) while preserving normal synaptic transmission.
Phencyclidine (PCP), dextromethorphan
NMDA antagonists with varying potency; relevant for toxicology and the pharmacology of dissociative states.
 |
|
|
|
|
|
|
|
|