|
|
|
|
|
|
|
-
-
Pharmacokinetics
-
Attribution:
-
Saffarzadeh A What is Pharmacokineticshttps://www.youtube.com/watch?v=CMRZqdrkCZw (8/2012)
-
Saffarzadeh Youtube Channel: https://www.youtube.com/channel/UCh93eqkE5oje6973bZXqM8A
-
-
-
-
-
"Absorption Overview"
-
Attribution:
-
Saffarzadeh A Drug Absorption Overview https://www.youtube.com/watch?v=eya9jR3v7i8 . (8/2012)
-
Areo Saffarzadeh Youtube Channel: https://www.youtube.com/channel/UCh93eqkE5oje6973bZXqM8A
-
-
-
-
Passive diffusion (aqueous or lipid environment): most common
-
Active transport: important for some drugs, particularly larger molecules.
-
-
-
Aqueous diffusion may occur:
-
Within large aqueous components (e.g.,interstitial space, cytosol)
-
Across epithelial membrane tight junctions
-
Across endothelial blood vessel lining
-
Through aqueous pores: allows diffusion of molecules with molecular weights up to 20,000 -- 30,000.
-
-
-
Driving force is the drug concentration gradient (described by Fick's Law ).
-
The driving force represents a tendency for molecules to move in the direction of higher concentration to lower concentration in accord with random molecular motion.
-
A traditional way of thinking about this is to imagine a fluid-filled container which is two sections divided by an imaginary plane.
-
The solution on one side is more concentrated in terms of some dissolved substance that is the solution on the other side of the boundary plane.
-
-
Recall that the molecules move randomly, suggesting that sometimes a molecule initially in the "low concentration" section can move to the "high concentration" section.
-
However, on balance, it is more likely that based on probability molecules will tend to move from the higher concentrations side to the lower concentrations side.
-
Suppose that initially there are 2,000 molecules on side A and 1,000 molecules on side B, a ratio of 2:1.
-
After a while we look again and find that there now are 1750 molecules on side A and 1250 molecules on side B.
-
A new ratio is been established (1750/1250 or 1.4 to 1), but the process continues until the ratio is approximately 1:1.
-
-
-
-
-
-
Fick's Law describes passive movement molecules down its concentration gradient.
Flux (J) (molecules per unit time) = (C1 - C2) · (Area ·Permeability coefficient) / Thickness
-
Where C1 is the higher concentration and C2 is the lower concentration
-
Area = area across which diffusion occurs
-
Permeability coefficient: drug mobility in the diffusion path
-
For lipid diffusion, lipid: aqueous partition coefficient -- major determinant of drug mobility
-
Partition coefficient reflects how easily the drug enters the lipid phase from the aqueous medium.
-
-
-
Thickness: length of the diffusion path
Katzung, B. G. Basic Principles-Introduction , in Basic and Clinical Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998, p 5.
-
-
-
Plasma protein-bound drugs cannot permeate through aqueous pores
-
Charged drugs will be influenced by electric field potentials {membrane potentials, important in renal, trans-tubular transport}
-
 II.
Lipid diffusion
II.
Lipid diffusion -
Lipophilic and Hydrophilic Drugs
-
Attribution:
-
AMBOSS: Medical Knowledge Distilled
-
AMBOSS: Medical Knowledge Distilled Youtube Channel:
-
-
-
Most important barrier for drug permeation due to:
-
Many lipid barriers separating body compartments
-
-
Lipid: aqueous drug partition coefficients described the ease with which a drug moves between aqueous and lipid environments
-
Ionization state of the drug is an important factor: charged drugs diffuse-through lipid environments with difficulty.
-
pH and the drug pKa, important in determining the ionization state, will influence significantly transport.
-
The pH and drug pKa determine the ratio of lipid-to aqueous-soluble forms for weak acids and bases as described by the Henderson-Hasselbalch equation.
-
Uncharged form: lipid-soluble
-
Charged form: aqueous-soluble, relatively lipid-insoluble (does not pass biological membranes easily)
-
-
-
Henderson-Hasselbalch equation: General form: log (protonated)/(unprotonated) = pKa-pH
-
For Acids: pKa = pH + log (concentration [HA] unionized)/concentration [A-]
-
note that if [A-] = [HA] then pKa = pH + log (1) or (since log(1) = 0), pKa = pH
-
-
For Bases: pKa = pH + log (concentration [BH+] ionized)/concentration [B]
-
note that if [B] = [BH+] then pKa = pH + log (1) or (since log(1) = 0), pKa = pH
-
-
The lower the pH relative to the pKa the greater fraction of protonated drug is found.
-
Recall that the protonated form of an acid is uncharged (neutral); however, protonated form of a base will be charged.
-
-
As a result, a weak acid at acid pH will be more lipid-soluble because it is uncharged and uncharged molecules move more readily through a lipid (nonpolar) environment, like the cell membrane, compared to charged molecules
-
Similarly a weak base at alkaline pH will be more lipid-soluble because at alkaline pH a proton will dissociate from molecule leaving it uncharged and thus free to move through lipid membrane structures
-
-
-
Lipid diffusion depends on adequate lipid solubility
-
 Drug ionization
(charged form) reduces a drug's ability to cross
a lipid bilayer.
Drug ionization
(charged form) reduces a drug's ability to cross
a lipid bilayer.
-
-
Drugs that are weak acids or bases
-
A weak acid is a neutral molecule that dissociates into an anion (negatively charged) and a proton (a hydrogen ion). For example:
-
C8H7O2COOH ⇄ C8H7O2COO- + H+
-
Neutral aspirin (C8H7O2COOH) in equilibrium with aspirin anion (C8H7O2COO- ) and a proton (H+ )
-
Weak acid: protonated form -- neutral, more lipid-soluble
-
-
Weak base: a neutral molecule that can form a cation (positively charged) by combining with a proton. Example:
-
C12H11CIN3NH3+ ⇄ C12H11CIN3NH2 + H+
-
Pyrimethamine cation (C12H11CIN3NH3+) in equilibrium with neutral pyrimethamine (C12H11CIN3NH2) and a proton (H+ )
-
Weak base: protonated form which is charged and therefore less lipid-soluble.
-
-
Examples:
-
Weak acids
pKa
Weak bases
pKa
-
Phenobarbital (Luminal)
7.1
-
Cocaine
8.5
-
Pentobarbital (Nembutal)
8.1
-
Ephedrine
9.6
-
Acetaminophen
9.5
-
Chlordiazepoxide (Librium)
4.6
-
Aspirin
3.5
-
Morphine
7.9
-
-
-
III. Special Carriers
-
Peptides, amino acids, glucose are examples of molecules then enter cells through special carrier mechanisms.
-
Carriers:
-
Active transport describes an energy requiring process which is saturable, meaning that transport is probably against the concentration gradient and involves a finite number of carriers, hence the process must be saturable when all carrier sites are filled.
-
Facilitated diffusion, while not requiring "energy" is also saturable (limited number of carrier sites)
-
Saturable (unlike passive diffusion) because of limited number of carrier sites--once those sites are filled, transport rates cannot be increased.
-
-
A property of carrier systems is that the process is subject to inhibition by other small molecules.
-
-
-
IV.
Endocytosis
and exocytosis:-
Entry into cells by very large substances (e.g., iron vitamin B12 , complexed with its binding protein moves across intestinal wall into the blood.
-
Neurotransmitter system examples for exocytosis:
-
Following neuronal electrical activation of nerve endings, two steps may be initiated:
-
Storage vesicles containing neurotransmitter fuse with cell membranes followed by
-
Release or diffusion of contents into the extracellular region.
-
-
-
-
Summary
-
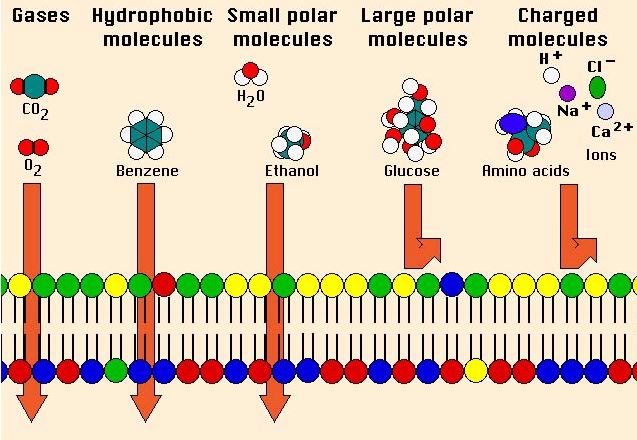
Figure Developed by Dr. Steve Downing, University of Minnesota
-
Stoelting, R.K., "Pharmacokinetics and Pharmacodynamics of Injected and Inhaled Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 1-17.
Dolin, S. J. "Drugs and pharmacology" in Total Intravenous Anesthesia, pp. 13-35 (Nicholas L. Padfield, ed), Butterworth Heinemann, Oxford, 2000
|
|
|
|
|
|
|